
 |
|
|||||||||||||||||||||||||||||||||
| [∞l±Ì‘u’ì] [±æÓê∆‰À˚Æa∆∑] [±æÓê∆‰À˚π©ë™…Ã] [ ’≤ÿ] | ||||||||||||||||||||||||||||||||||
| ‰N €…ã∫ ±±æ©øµùô’\òI…˙ŒÔø∆ºº”–œÞπ´Àæ | ≤Èø¥‘ìπ´ÀæÀ˘”–Æa∆∑ >> |
°æÿõÃñ∫Õ“é∏Ò°øT115-01£¨20 rxn
°æÆa∆∑∏≈ ˆ°ø
ÎSôCÕª◊É «ÍU ˆµ∞∞◊Ÿ|ΩYòã∫Õπ¶ƒÐ÷ÆÈgµƒÍPœµ°¢∏ƒþMµ∞∞◊Ÿ|–‘ƒÐµƒ÷ÿ“™π§æþ°£StarMutÎSôCÕª◊É‘áÑ©∫–ª˘”⁄“◊ÂePCR (error-prone PCR) ºº–g£¨¿˚”√Taq DNA polymerase≤ªæþ”–3°‰°˙5°‰–£å¶π¶ƒÐµƒÃÿ–‘£¨‘⁄Ãÿ∂®µƒ∑¥ë™æèõ_Ûwœµ÷–£¨œÚîU‘ˆµƒƒøµƒª˘“Ú÷–“˝»ÎÎSôCÕª◊É√Ð¥a◊”°£éß”–ÎSôCÕª◊ɵƒîU‘ˆÆaŒÔÕ®þ^Îp√∏«–£¨þBΩ”µΩ±Ìþ_ðdÛw÷–òãΩ®ŒƒéÏ£¨»ª∫ÛÞDªØ»Î±Ìþ_ÀÞ÷˜÷–£¨þM––µ∞∞◊ªÓ–‘∫Yþx°£»Áπ˚Ωõ“ª¥ŒÕª◊É∑¥ë™≤ªƒÐ´@µ√ùM“‚µƒΩYπ˚£¨ø…≤…”√þB¿m“◊ÂePCR (sequential error-prone PCR)≤þ¬‘£¨º¥å¢“ª¥ŒPCRîU‘ˆµ√µΩµƒ”–”√Õª◊ɪ˘“Ú◊˜ûÈœ¬“ª¥ŒPCRîU‘ˆµƒƒ£∞£¨þB¿m∑¥èÕµÿþM––ÎSôC’T◊É£¨ π√ø“ª¥Œ´@µ√µƒÕª◊É¿€∑e∂¯Æa…˙∏¸”–“‚¡xµƒÕª◊É°£
±æ‘áÑ©∫–∞¸∫¨”–ÉûªØµƒ2 x StarMut Random System°¢StarMut Enhancer∫ÕddH2O»˝∑NΩM∑÷£¨ π”√ïr÷ª–˺”»Îþm¡øµƒDNAƒ£∞Â∫Õ∫œ≥…µƒÉ…ólîU‘ˆ“˝ŒÔ£¨≤¢”√ÀÆ—a◊„Ûw∑e£¨º¥ø…þM––îU‘ˆ∑¥ë™£¨≤Ÿ◊˜∫ܱ„øÏÀŸ£¨¥Û¥Ûúp…Ÿ¡À∂ý¥Œº”ò”ø…ƒÐ‘Ï≥…µƒ≥ˆÂe∫ÕŒ€»æôCï˛°£2 x StarMut Random System∫¨”–ÉûªØù‚∂»µƒTaq DNA polymerase°¢dNTPs°¢∑¥ë™æèõ_“∫∫Õ∑Ä∂®Ñ©£¨ø…◊Ó¥ÛœÞ∂»µÿøÀ∑˛≥£“é“◊ÂePCR ºº–g÷–£¨”…”⁄Taq DNA polymerase±æ…̵ƒ∆´ê€–‘‘Ï≥…µƒ“‘GCÕª◊ÉûÈ÷˜µƒ»±¸c£¨´@µ√œýå¶æ˘∫‚µƒÕª◊É◊V°£âAª˘Õª◊ɬ ø…Õ®þ^þm¡øÃ̺”StarMut Enhancer°¢’{’˚ƒ£∞ÂDNA¡ø∫Õ∏ƒ◊ÉPCRîU‘ˆ—≠≠hîµþM––øÿ÷∆°£
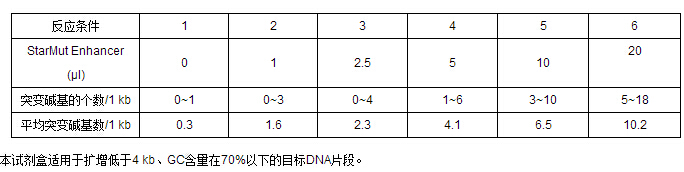
œ¬±Ì¡–≥ˆ“‘10 ngŸ|¡£DNAûȃ£∞£¨PCRîU‘ˆ“ªól1 kb DNA∆¨∂Œ(20ÇÄ—≠≠h)∫Ûúy–Úôzúyµ√µΩµƒÕª◊ɬ ΩYπ˚°£–Ë“™◊¢“‚µƒ «£¨”…”⁄≤ªÕ¨DNAƒ£∞µƒâAª˘ΩM≥…≤ªÕ¨°¢ÈL∂»≤ª“ª£¨“‘º∞≤ªÕ¨“˝ŒÔµƒîU‘ˆ–߬ ¥Ê‘⁄≤ÓÆ꣨À˘“‘º¥ π‘⁄œýÕ¨µƒPCR∑¥ë™ólº˛œ¬£¨É…ΩMPCRÆaŒÔÀ˘µ√µΩµƒÕª◊ɬ “≤ø…ƒÐ≤ªÕ¨°£“Ú¥ÀŒ“ÇÉΩ®◊h∏˘ì˛åçÚûµƒæþÛw“™«Û£¨ ◊œ»þM––∂ýÇÄ–°Ûwœµ(20 ¶Ãl)îU‘ˆÓAåçÚû£¨∑÷Ñeº”»Î≤ªÕ¨Ûw∑eµƒStarMut Enhancer(»Á0 ¶Ãl°¢1¶Ãl°¢5 ¶Ãl°¢10 ¶Ãlµ»)£¨√˛À˜≥ˆ∑˚∫œƒøòÀÕª◊ɬ µƒ∑¥ë™ólº˛∫Û£¨‘Ÿ∑≈¥ÛîU‘ˆÛwœµ°£∆‰À˚”∞ÌëÕª◊ɬ µƒ“ÚÀÿ“ä°æ◊¢“‚ ¬Ìó°ø°£
°æÆa∆∑ΩM∑÷°ø
2 x StarMut Random System
0.5 ml
StarMut Enhancer
0.1 ml
ddH2O
1 ml
°æ±£¥Êólº˛°ø
−20°Ê±£¥Ê£¨”––ß∆⁄“ªƒÍ£ªΩõ≥£ π”√£¨ø…”⁄4°Ê±£¥Ê£¨”––ß∆⁄»˝ÇÄ‘¬°£
°æ π”√∑Ω∑®°ø
1£Æ“˝ŒÔ‘O”ã‘≠Ñt£∫
(1) ’˝°¢∑¥œÚîU‘ˆ“˝ŒÔ∏˜“ªól£¨ÈL∂»ºs20~45ÇÄâAª˘£¨3°Ø∂À∑÷Ñe≈cƒøòÀÕª◊ÉDNA∆¨∂Œµƒ…œœ¬”ŒΩY∫œ£ª
(2) ±M¡ø墓˝ŒÔµƒGC∫¨¡øøÿ÷∆‘⁄40~60%£ª
(3) “˝ŒÔ»Áéß”–øÀ¬°√∏«–Œª¸c£¨±ÿÌöÃ̺”◊„âÚµƒ±£◊oâAª˘“‘¥_±£√∏«––߬ £ªåçÚû◊C√˜£¨“˝ŒÔΩY∫œ‘⁄ƒøòÀDNA∆¨∂Œµƒ√∏«–Œª¸cÕ‚Ç»ø…÷∏þ√∏«––߬ £¨‘ˆº”ÞDªØµƒøÀ¬°îµ¡ø°£
2£ÆÎSôCÕª◊É∑¥ë™£∫
(1) PCR∑¥ë™Ûwœµ£∫
œÚPCR±°±⁄πÐ÷–“¿¥Œº”»Îœ¬¡–‘áÑ©
Ÿ|¡£DNAƒ£∞Â(1~10 ng/¶Ãl) * 1 ¶Ãl
2 x StarMut Random System 25 ¶Ãl
’˝œÚîU‘ˆ“˝ŒÔ (10 ¶ÃM) 1 ¶Ãl
∑¥œÚîU‘ˆ“˝ŒÔ (10 ¶ÃM) 1 ¶Ãl
StarMut Enhancer 0~20 ¶Ãl
ddH2O —a◊„Ûw∑e÷¡ 50 ¶Ãl
(2) ªÏÑÚ∫Û∂Ãï∫Îx–ƒ£¨∑≈»ÎPCRÉx°£
(3) PCR—≠≠h֢ƒ‘O÷√£∫
95°Ê 2 min
94°Ê 30 sec
50~65°Ê 1 min x 16~25 cycles*
72°Ê 1 min/1 kb
72°Ê 7 min
*Õª◊ɬ ø…Õ®þ^∏ƒ◊É∆ ºƒ£∞Âù‚∂»∫ÕîU‘ˆ—≠≠hîµþM––øÿ÷∆°£∆ ºƒ£∞Âù‚∂»‘Ω∏þ£¨Õª◊ɬ ‘ΩµÕ£ªîU‘ˆ—≠≠hΩ∏þ£¨Õª◊ɬ ‘Ω∏þ°£
3£Æ»°1~5 ¶Ãl PCRÆaŒÔÎä”æôzúyóléßù‚∂»∫ÕÃÿÆê–‘°£
4£Æ £”ýµƒPCRÆaŒÔÎä”棨«–ƒzªÿ ’ƒøòÀDNA∆¨∂Œ°£
5£Æ√∏«–°¢þBΩ”°¢ÞDªØµΩ±Ìþ_ÀÞ÷˜æ˙÷Í÷–þM––∫Yþx°£
°æ◊¢“‚ ¬Ìó°ø
1£Æ ”…”⁄≤ªÕ¨DNAƒ£∞µƒâAª˘ΩM≥…≤ªÕ¨°¢ÈL∂»≤ª“ª£¨“‘º∞≤ªÕ¨“˝ŒÔµƒîU‘ˆ–߬ ¥Ê‘⁄≤ÓÆ꣨À˘“‘º¥ π‘⁄œýÕ¨µƒPCR∑¥ë™ólº˛œ¬£¨É…ΩMPCRÆaŒÔÀ˘µ√µΩµƒÕª◊ɬ “≤ø…ƒÐ≤ªÕ¨°£“Ú¥ÀŒ“ÇÉΩ®◊h∏˘ì˛æþÛwåçÚû“™«Û£¨ ◊œ»þM––∂ýÇÄ–°Ûwœµ£®20 ¶Ãl£©îU‘ˆÓAåçÚû£¨∑÷Ñeº”»Î≤ªÕ¨Ûw∑eµƒStarMut Enhancer£®»Á0 ¶Ãl°¢1 ¶Ãl°¢5 ¶Ãl°¢10 ¶Ãlµ»£©£¨Õ®þ^úy–ÚªÚªÓ–‘ôzúyµ»∑Ω∑®£¨√˛À˜≥ˆ∑˚∫œƒøòÀÕª◊ɬ µƒ∑¥ë™ólº˛∫Û£¨‘Ÿ∑≈¥ÛîU‘ˆÛwœµ°£
2£Æ ∆ ºDNAƒ£∞µƒù‚∂»å¶Õª◊ɬ ”–∫Ð¥Û”∞Ì루ծ≥£ø…Õ®þ^÷∏þªÚΩµµÕDNAƒ£∞µƒù‚∂»ÅÌ’{’˚Õª◊ɬ °£Ëb”⁄≤ªÕ¨–ÕÃñµƒ∑÷π‚π‚∂»”ãôzúyµƒDNAù‚∂»¥Ê‘⁄∆´≤Ó£¨DNAƒ£∞Â◊Ó∫√‘⁄√∏«–æÄ–‘ªØ∫Û£¨≤…”√ƒ˝ƒzÎä”æ∑Ω∑®£¨≈c“—÷™ù‚∂»µƒæÄ–‘ªØÎpÊúDNAªÚ…Ã∆∑ªØµƒDNA markerþM––嶱»£¨¥_∂®∆‰ù‚∂»°£
3£Æ Õª◊É∑¥ë™ÆaŒÔ±ÿÌöþM––«–ƒzªÿ ’Ãé¿Ì£¨»•≥˝DNAƒ£∞°¢PCRÆaŒÔ…œΩY∫œµƒTaq√∏“‘º∞∆‰À˚ÎsŸ|°£≥£“鵃““¥º≥¡µÌ°¢π˃zƒ§£®÷È£©ªÚ≤£¡ßƒÃŒ¸∏Ωµ»∑Ω∑®£¨æ˘üo∑®»•≥˝ΩY∫œµƒTaq√∏£¨∫Û’þø…ƒÐ’⁄±Œ√∏«–Œª¸c£¨”∞ÌëøÀ¬°–߬ °£
4£Æ Ω®¡¢ÎSôCÕª◊ÉŒƒéÏÕ®≥£–Ë“™10~200 ng/¶Ãl £®œýÆî”⁄500 ng ~10 ¶Ãg/50 ¶ÃlÛwœµ£©µƒPCRÆaŒÔ°£»Á”ˆÆa¡ø≤ª◊„£¨ø…Õ®þ^œ¬¡–∑Ω∑®Ã·∏þÆa¡ø£∫
(1) Õª◊ɬ ∑˚∫œ–Ë«Ûïr£¨ø…∑≈¥ÛPCRÛwœµ£¨ªÚ«–ƒzªÿ ’PCRÆaŒÔ∫Û£¨≤…”√≥£“éPCR∑¥ë™ólº˛þM––îU‘ˆ£ª
(2) Õª◊ɬ µÕ”⁄–Ë«Ûïr£¨Ã·∏þPCRîU‘ˆ—≠≠hîµ£¨ªÚ«–ƒzªÿ ’PCRÆaŒÔ∫Û£¨þM––µ⁄∂˛ðÜÎSôCÕª◊É∑¥ë™£ª
(3) ÷ÿ–¬‘O”ãîU‘ˆ“˝ŒÔ;
(4) ΩµµÕÕÀªúÿ∂»£ª
(5) ¥_±£ƒ£∞Ÿ|¡ø£¨≤…”√ƒ˝ƒzÎä”æ∑Ω∑®æ´¥_∂®¡ø°£
5£Æ 嶔⁄éß”–øÀ¬°√∏«–Œª¸cµƒDNAƒ£∞£¨±ÿÌö‘⁄PCR∑¥ë™ΩY ¯∫Û£¨ π”√DpnIÕÍ»´œ˚ªØ«Â≥˝º◊ª˘ªØµƒƒ£∞£¨‘Ÿ«–ƒzªÿ ’ƒøòÀDNA∆¨∂Œ°£å¶”⁄∑«º◊ª˘ªØµƒŸ|¡££®¿˝»Á胥ۃcóUæ˙JM110ªÚSCS110æ˙÷Í÷–÷»°µƒŸ|¡££©£¨ø…Õ®þ^ÞDªØdam£´µƒ¥ÛƒcóUæ˙æ˙÷Í£®»ÁDH5¶¡°¢TOP10°¢JM109°¢XL1-Blueµ»£©£¨‘Ÿ≥È÷´@µ√º◊ª˘ªØµƒŸ|¡£◊˜ûÈPCR∑¥ë™ƒ£∞°£
6£Æ Ωõþ^Îp√∏«–µƒøÀ¬°ðdÛw‘⁄≤»ÎÕª◊ÉDNA∆¨∂Œ«∞£¨ë™ ◊œ»Õ®þ^◊‘…ÌþBΩ”ôzúy£¨¥_±£òOµÕµƒ◊‘þB±≥æ∞°£±ÿ“™ïrø…≤…”√»•¡◊À·ªØ°¢«–ƒzªÿ ’µ»∑Ω∑®∞—◊‘þB¬ Ωµ÷¡◊ÓµÕ£¨“‘√‚”∞Ìë∫Û¿mþBΩ”∑¥ë™°£
°ælj◊¢°ø
±æÆa∆∑ÉHπ©ø∆—– π”√°£‘⁄¥_’JÆa∆∑Ÿ|¡ø≥ˆ¨FÜñÓ}ïr£¨±æπ´Àæ≥–÷ZûÈøÕëÙ√‚ŸM∏¸ìQµ»¡øµƒŸ|¡ø∫œ∏ÒÆa∆∑°£‘⁄À˘”–«Èõrœ¬£¨±æπ´Àæ嶥ÀÆa∆∑À˘≥–ì˙µƒÿü»ŒÉHœÞ”⁄¥ÀÆa∆∑µƒÉr÷µ±æ…Ì°£

- MCE÷–á¯å¢îy‘áÑ©Æa∆∑¡¡œý√¿á¯÷•º”∏Á2025 AACRƒÍï˛
- 2024ƒÍ"MCE÷–ᯅ˙√¸ø∆åW—–æø¥ŸþM™Ñ"‘u™ÑΩYπ˚π´≤º
- Ã’–g…˙ŒÔ÷±≤•ÓA∏Ê£∫ ÐÛwÀé¿ÌåW≈cÀéŒÔ∞l¨F—–æø
- ∞≤ΩðÇê√‚“þΩMªØ∑®PD-L1ôzúy‘áÑ©∫–´@µ√öW√ÀIVDR’J◊C
- ÃϬ°◊‘÷˜—–∞lHCV∫ÀÀ·ôzúy‘áÑ©∫–´@≈˙Àé±Oæ÷»˝ÓêNMPA
- µ¬á¯Massive PhotonicsÕ∆≥ˆ∂ýøÓDNA-PAINT∏˜œµ‘áÑ©∫–
- IPHASE—˚ƒ˙π≤∏∞BIOCHINA2025µ⁄ Æå√“◊ŸQ…˙ŒÔÆaòI¥Ûï˛
- Ãm≤©¿˚µ¬Õ∆≥ˆ–¬∆∑∏þ±£’ÊøÏÀŸîU‘ˆ PCR mix‘áÑ©∫–
- OK432‘⁄ƒ[¡ˆ∫Õ√‚“þ—–æø÷–µƒ◊˜”√ôC÷∆º∞ë™”√ÉûÑð
- ËF’{ÀÿHepcidin-25◊˜ûÈËF¥˙÷x∫Õ√‚“þ∑¥ë™ÍPÊI’{πù“Ú◊”µƒπ¶ƒÐ∫Õ◊˜”√
- ëµ∞∞◊Fibronectin‘⁄ºö∞˚≈ýB÷–µƒπ¶ƒÐº∞◊˜”√
- TDP-43µƒΩYòã≈cπ¶ƒÐº∞‘⁄ùuɈ∞Y(ALS)µ»…ÒΩõÕÀ–––‘º≤≤°÷–µƒ◊˜”√ôC÷∆
- DNP-BSA(2,4-∂˛œıª˘±Ω≈º¬ì≈£—™«Â∞◊µ∞∞◊)‘⁄√‚“þåW—–æø≈côzúy÷–µƒ◊˜”√
- AbMole LPS£®÷¨∂ý룩‘⁄√‚“þº§ªÓ°¢Ñ”ŒÔ‘σ£µ»∑Ω√ʵƒë™”√º∞∞∏¿˝∑÷œÌ
- AbMoleµ∞∞◊√∏“÷÷∆Ñ©Cocktail‘⁄WBåçÚû÷–µƒë™”√ÉûÑðº∞∞∏¿˝∑÷œÌ
- AbMole÷ÿΩMCRM197ðdÛwµ∞∞◊µƒÃÿ–‘º∞‘⁄“þ√Á—–∞l÷–µƒë™”√