
 |
分子對接是依據(jù)配體與受體作用的“鎖-鑰原理”,模擬配體小分子與受體生物大分子相互作用的一種技術(shù)方法。配體與受體相互作用是分子識別的過程,主要包括靜電作用、氫鍵作用、疏水作用、范德華作用等
|
|||||||||||||||||
| [發(fā)表評論] [本類其他服務(wù)] [本類其他服務(wù)商] | ||||||||||||||||||
| 服務(wù)商: 北京中大唯信科技有限公司 | 查看該公司所有服務(wù) >> |
分子對接能有效運用于: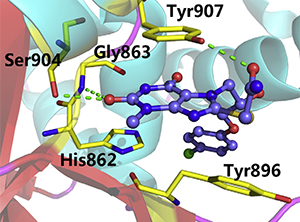
- 探索藥物小分子和大分子受體的具體作用方式和結(jié)合構(gòu)型;
- 篩選可以與靶點結(jié)合的先導(dǎo)藥物;
- 解釋藥物分子產(chǎn)生活性的原因;
- 指導(dǎo)合理地優(yōu)化藥物分子結(jié)構(gòu)。
【服務(wù)內(nèi)容】
- 同源模建(受體無結(jié)構(gòu))
- 受體、配體準備
- 結(jié)合位點判斷
- 蛋白柔性構(gòu)象探索
- 配體構(gòu)象數(shù)據(jù)庫準備
- 分子對接
- 結(jié)果分析、評價
隨著X-Ray 衍射和NMR 技術(shù)的發(fā)展,已經(jīng)有大量的靶標蛋白的三維晶體結(jié)構(gòu)被解析出來,得到蛋白的晶體結(jié)構(gòu)后并不能直接用于分子對接。實際上,在蛋白的解析過程中經(jīng)常會有各種各樣的錯誤,比如原子的缺失、蛋白的二級序列和三維結(jié)構(gòu)不能對應(yīng)等等,這些都會影響對接的準確性,特別是當(dāng)這些錯誤出現(xiàn)在配體的結(jié)合口袋內(nèi)時。因此,在對接前必須對這些錯誤進行更正。無論是X-Ray 還是NMR 都只能確定重原子的位置而沒有氫原子的位置信息,在對接前就需要先進行加氫質(zhì)子化,標示局部電性,這樣才能用于對接。
準備蛋白結(jié)構(gòu)后需要尋找藥物分子結(jié)合的活性位點,而蛋白表面的拓撲非常復(fù)雜、多樣,物理化學(xué)性質(zhì)也異常多樣化,究竟哪些位點才是小分子藥物結(jié)合的位置,并能抑制或激活蛋白的活性呢?實際上針對靶標蛋白必然有一些相應(yīng)的研究和其生物學(xué)功能的注釋。
在大多數(shù)情況下,蛋白也是通過結(jié)合天然的配體(大分子或小分子)來發(fā)揮其生物學(xué)功能。這些天然配體的結(jié)合位點很可能就是其抑制劑或激動劑的結(jié)合位點。如果缺少這些相應(yīng)的生物學(xué)注釋,也可以借助于計算分析,從拓撲、物化性質(zhì)等多個角度來考察蛋白表面,找到合適的結(jié)合位點,并和實驗信息相結(jié)合,最后確定活性位點。
眾所周知,蛋白-配體相互作用過程中,存在誘導(dǎo)契合效應(yīng),結(jié)合過程中其構(gòu)象都會發(fā)生相應(yīng)的變化,一個準確的對接必須考慮到受體和配體的柔性。現(xiàn)在的軟件工具雖然很多都宣稱可以進行受體柔性對接,但是方法上都有比較大的局限性,可能只是通過力場優(yōu)化等方法進行側(cè)鏈的構(gòu)象優(yōu)化。
我們可以通過計算機模擬的方法考察蛋白可能存在的幾種不同的構(gòu)象,通過這些構(gòu)象作為對接的起點能更多的考慮蛋白的柔性。另一個柔性是配體的柔性。盡管在對接過程中軟件會自動的考慮配體的柔性,比如旋轉(zhuǎn)一些可旋轉(zhuǎn)鍵。但是這種構(gòu)象的產(chǎn)生也是比較有限的,比如無法充分考慮到飽和環(huán)的構(gòu)象。我們可以通過構(gòu)象搜索、飽和環(huán)構(gòu)象搜索等方法盡可能的遍歷配體的優(yōu)勢構(gòu)象來作為對接的構(gòu)象庫,從而提高準確性!對接后一般通過結(jié)合自由能的打分來進行排序。可能每個配體有多種的結(jié)合構(gòu)象,我們通過綜合的評價方法來挑選最有可能的結(jié)合模式,比如結(jié)合自由能的打分、分子的應(yīng)力能等,并且結(jié)合人為的判斷挑選來找到真正合理的結(jié)合方式。
- BSI邀您參加蛋白質(zhì)組學(xué)數(shù)據(jù)分析培訓(xùn)會
- 單細胞空間多組學(xué)技術(shù)論壇暨廣州站生信培訓(xùn)班報名中
- SBC單細胞及空間多組學(xué)實驗與生信分析培訓(xùn)班招生
- 小海龜發(fā)布百個橫向課題、千篇SCI-生命科學(xué)合作計劃
- 空間多組學(xué)研究策略及生信分析培訓(xùn)班火熱招生中
- SBC ToolBox云平臺VIP專區(qū)再添新模塊GSEA數(shù)據(jù)分析
- SBC芯云講壇:國家自然科學(xué)基金的準備和申報技巧
- 第十期SBC單細胞及空間轉(zhuǎn)錄組測序培訓(xùn)班報名通知
- 非負矩陣分解NMF算法助力單細胞轉(zhuǎn)錄組數(shù)據(jù)分析
- 深度神經(jīng)網(wǎng)絡(luò)助力對腫瘤細胞鑒定及空間惡性區(qū)域的識別
- 腫瘤純度和倍性評估工具Sequenza的安裝和使用方法
- 單細胞高級分析百篇文獻結(jié)果展示匯總(六)
- 單細胞高級分析百篇文獻結(jié)果展示匯總(五)
- 使用SBC ToolBox 單細胞抖動圖模塊美化差異marker基因
- SBC ToolBox在Small RNA-seq定量數(shù)據(jù)分析中的應(yīng)用(下)
- SBC ToolBox在Small RNA-seq定量數(shù)據(jù)分析中的應(yīng)用(上)
 瀏覽該公司同類服務(wù)
瀏覽該公司同類服務(wù)