
高通量蛋白質組學通過大規模樣本集推進臨床研究
隨著蛋白質組學技術應用的快速發展,離子淌度這一概念的引入,蛋白質組學已步入4D新時代,引領臨床蛋白質組學在鑒定準度、鑒定深度、定量準確性、檢測周期等性能上全面提升,并在基礎研究、轉化醫學、臨床蛋白質組學等各個方面展現出了廣闊的發展前景和強大的生命力。值得一提的是,在新冠病毒疫情全球大流行之際,4D蛋白質組學下的大隊列新冠患者樣本臨床蛋白質組學研究也為抗擊病毒做出了貢獻。
4D蛋白質組學究竟是如何輕松解決大隊列、復雜體系以及微量樣本檢測的?而高通量蛋白質組學又是如何通過大規模樣本集推進臨床研究的?帶著這些疑問,一起來看看臨床蛋白質組學專家是如何利用4D蛋白質組學推進臨床研究的。
“我們的研究所與醫院緊密相連,有許多合作,其中蛋白質組學已成為主要研究戰略的一部分。這意味著臨床研究人員對其樣本進行蛋白質組學分析的需求越來越大。與此同時,每次臨床研究的平均樣本數也大幅上升。而這正是高通量蛋白質組學大顯身手的地方。”
--牛津大學標靶研究所臨床蛋白質組學Roman Fischer教授
1
臨床蛋白質組學面臨的挑戰
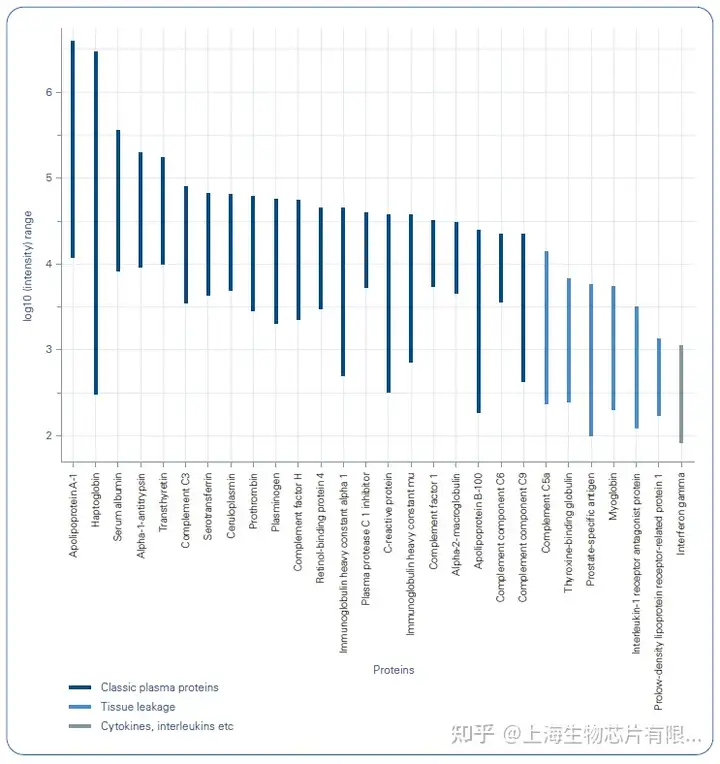
過去,蛋白質組學并不是臨床研究的驅動力,因為樣本集通常非常小。此外,由于當時質譜儀的速度、靈敏度和分辨率有限,很難實現完整蛋白質組的覆蓋。臨床樣本通常具有挑戰性,蛋白質含量極高,動態范圍高[1]。這給質譜分析帶來了復雜性,因為質譜儀的動態范圍通常比臨床樣本中蛋白質的動態范圍小得多[2]。
而基于Bruker timsTOF Pro的4D蛋白質組學的出現,改變了這種情況,提高了分析時間和穩健性,使高通量蛋白質組學成為可能。

基于timsTOF Pro的4D蛋白質組學使研究人員能夠重復測量所有檢測到的離子的碰撞截面(CCS)值,這些值可用于進一步提高系統的選擇性,從而從復雜樣本和短梯度分析中獲得越來越可靠的相對定量信息[3]。
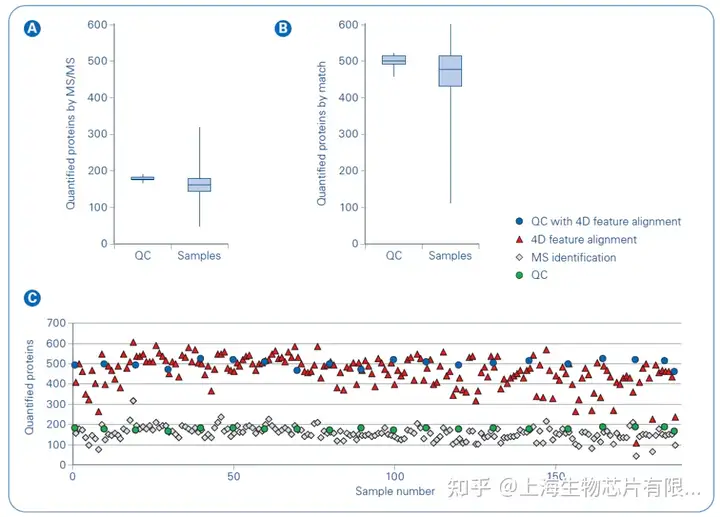
有研究團隊發現,基于timsTOF Pro的4D蛋白質組學除了能夠進行離子遷移信息的常規測量,還在保留時間、質荷比(m/z)、離子遷移率 (1/K0) 和MS1強度方面實現高質量的4D特征校準(圖1)。校準提高了定量血漿蛋白的數量,在一次11.5分鐘的LC-MS/MS運行中超過500個蛋白質,如果將所有運行結合起來,共定量772個蛋白質(圖2)[4]。達到這種深度提供了全新的可能性,分析大樣本隊列的數百至數千個樣本,以發現血漿中的生物標志物。
PASEF技術可以在不損失靈敏度或分辨率的情況下實現>100 Hz的序列速度。這是通過使四極隔離質量窗口與TIMS通道中特定肽包的洗脫時間同步來實現的。通過多次選擇,可以提高低豐度肽的MS/MS光譜質量。從少于200 ng的樣品負載中可以獲得更好的結果,因此減少了樣品制備成本和MS維護頻率。使用90分鐘的梯度長度,可以從典型的人類細胞系裂解液中識別出5400多個蛋白質組。
穩健性是基于timsTOF Pro的4D蛋白質組學研究的另一個主要優勢。關于這種能力對蛋白質組學研究的影響,Fischer教授是這樣說的:“通常情況下,一個儀器需要在200個血樣后進行清潔。在高通量環境下,這意味著高頻率的儀器清潔,嚴重影響研究進度。而我們在timsTOF Pro儀器上運行的最大樣本批次是4500次非衰竭血液注射,這是極具挑戰性的樣本之一。
基于timsTOF Pro的4D蛋白質組學還可以從少量高度復雜的混合物中提供可重復的定量信息。Fischer教授解釋道:“達到100+Hz,這意味著它有可能每秒識別100個或更多的肽。與其他儀器相比,我們可以真正縮短梯度時間,即使是對于總細胞裂解物之類的樣本,也不會影響我們獲得的數據深度。此外,該儀器也非常敏感。可以說,是在不會影響靈敏度的前提下提高了速度和穩健性。”
2
對高通量蛋白質組學的需求不斷增長
自從越來越多的人將蛋白質組學應用到課題研究中以來,需求大幅上升,這是對其他組學的補充。
Fischer教授和他的團隊使用蛋白質組學來確定腫瘤在空間環境中的功能。他說道:“因為現在我們可以將蛋白質組學與其他技術相結合,如激光捕獲顯微切割(LMD),這是一種非常有趣的方法,因為它允許我們維持蛋白質組的空間背景。目前,我們仍在努力了解大型結構(如腫瘤或器官)中的空間分辨分子相互作用。我們在腫瘤血管周圍看到炎癥標記物,但在其他地方看不到,因此我們試圖了解這些腫瘤在分子水平上發生了什么。這將影響靶向蛋白質以及如何將藥物輸送到最有效的位置。通過結合MS和LMD,我們可以重新定義表型。基本上,蛋白質組成為細胞表型的一部分,因為它現在可以通過空間分辨率觀察到。我喜歡稱之為‘表型蛋白質組’。顯然,這種方法也可以以空間對應的方式與基因組或其他組學分析相結合。”
3
新冠肺炎患者的分類
當前,基于timsTOF Pro的4D蛋白質組學最值得一提的應用,就是實現新冠肺炎感染中宿主免疫反應的深層表型[5]。
這項研究涉及200-300名患者,其中一些患者在多個時間點采集樣本,并在Bruker-timsTOF Pro上使用高通量蛋白質組學進行分析。該研究采用了一種新的方法,使用這些樣本的亞組與其他組學技術進行分析,例如某些血細胞的位點分析。血漿蛋白質組能夠將亞表型分型到患者群中,這可以預測嚴重程度和結果(圖3和4)[5]。在對同一組樣本進行的這項大型研究中使用的8種組學中,蛋白質組學最適合將新冠肺炎患者分為不同的疾病嚴重程度組,而其他技術則提供了數據來幫助理解潛在的生物學意義。
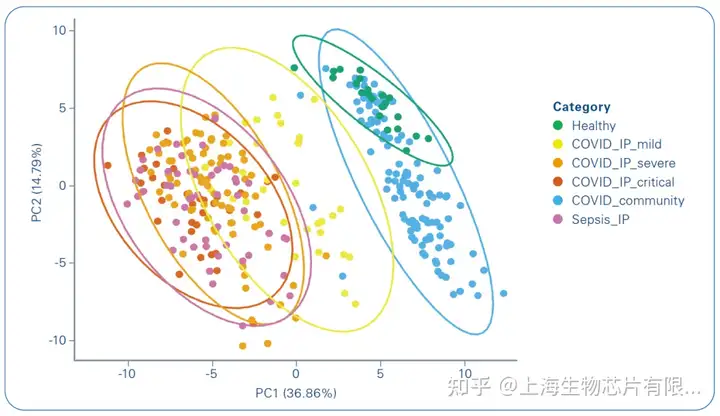
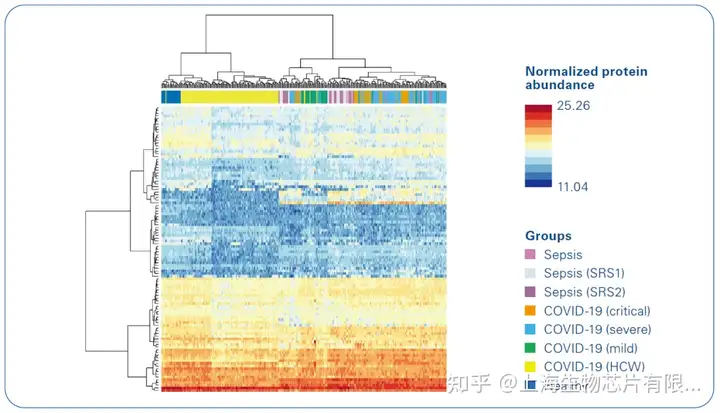
該蛋白質組分析確定了特定的血漿急性期蛋白水平作為嚴重疾病的指標,并有證據表明急性期炎癥、補體激活/攻擊、纖維蛋白凝塊、蛋白酶、血清淀粉樣蛋白、組織壞死、受體介導的內吞作用和膽固醇轉運。發現了血漿蛋白特征,可用于將急性住院新冠肺炎病例分層為疾病亞表型,聚類成員可為反應狀態提供信息,并與差異28天死亡率相關。
這一多組學血液圖譜等研究將為新冠肺炎未來的藥物開發、臨床試驗設計和個性化藥物治療方法提供必要的見解。
參考文獻:
[1]Cox J and Mann M (2011), Quantitative, High-Resolution Proteomics for Data-Driven Systems Biology, Annu. Rev. Biochem. 80: 273-299.
[2]Keshishian H, Burgess MW, Specht H, Wallace L, Clauser KR, Gillette MA, & Carr SA. (2017), Quantitative, multiplexed workflow for deep analysis of human blood plasma and biomarker discovery by mass spectrometry. Nature protocols, 12(8), 1683-1701.
[3]Meier F, Brunner AD, Koch S, Koch H, Lubeck M, Krause M, Goedecke N, Decker J, Kosinski T, Park M, Bache N, Hoerning O, Cox J, Räther O, Mann M. (2018), Online parallel accumulation – serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer, Mol. Cell. Proteomics, https://doi.org/10.1074/mcp. TIR118.000900.
[4]Kosinski T, Heilig R, Bensaddek D, Bache N, Bjeld Hørning O, Fischer R, Koch H., Plasma proteomics goes high throughput – timsTOF Pro with PASEF and 4D feature alignment to quantify 500 plasma proteins in 11.5 min. Bruker Daltonics 03-2019, LCMS-151, 1867805.
[5]Covid-19 Multi-omics Blood ATlas (COMBAT) Consortium (2022), Cell 185, 916–938, 2022. Published by Elsevier Inc. https://doi.org/10.1016/j.cell.2022.01.012 ll



