
利用磷酸化組和代謝組揭示膿毒癥中EGFR胞內轉運機制

敗血癥是指機體對病原體感染的先天免疫反應失調,可導致組織損傷,甚至危及生命。巨噬細胞的持續激活可能是加速膿毒癥反應的機制。而巨噬細胞中的代謝級聯反應被認為是其激活的特征。EGFR(表皮生長因子受體)是一種跨膜受體酪氨酸激酶,可與TLR4(Toll 樣受體 4)共同調節內毒素血癥中巨噬細胞的激活,EGFR的磷酸化增加了TLR4細胞表面表達和信號轉導。然而,巨噬細胞中EGFR在LPS(脂多糖)作用下的胞內轉運過程及其生理意義尚不清楚。
2022年11月,廣東醫科大學附屬醫院麻醉科唐靖課題組與解放軍南部戰區總醫院劉志峰課題組合作在Cell Death & Disease(IF9.685 )期刊上發表了題為“EGFR tyrosine kinase activity and Rab GTPases coordinate EGFR trafficking to regulate macrophage activation in sepsis”的文章,該研究利用磷酸化修飾組和代謝組學等方法發現LPS可增加巨噬細胞表面EGFR的表達,Rab10有助于EGFR的質膜運輸。Rab5a介導早期EGFR內吞,而EGFR/MAPK14/Rab7a調節晚期EGFR內吞。此外,抑制細胞表面EGFR表達降低M1極化,并通過激活ppar γ介導的谷氨酰胺代謝促進M2極化。 最后,抑制EGFR磷酸化使巨噬細胞的平衡從M1向M2傾斜,并減弱了LPS誘導的炎癥和組織損傷。中科新生命提供了磷酸化修飾組和非靶向代謝組技術服務支持。
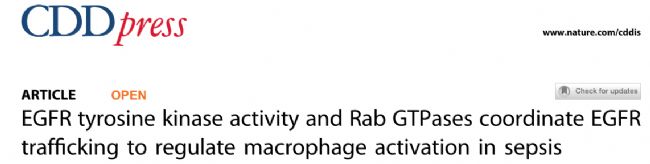
步驟1:LPS促進巨噬細胞表面EGFR的激活和表達;
步驟2:EGFR/MAPK14/Rab7a(S72)調節晚期EGFR內吞;
步驟3:Rab5a介導EGFR在巨噬細胞中的早期內化;
步驟4:Rab10促進EGFR從高爾基體運輸到細胞表面;
步驟5:抑制EGFR磷酸化可抑制巨噬細胞中M1極化、通過激活PPARγ調節谷氨酰胺代謝促進M2極化;
步驟6:抑制EGFR磷酸化可使M1表型轉變為M2表型,減輕膿毒癥引起的急性肺損傷。
研究結果
1. LPS促進巨噬細胞表面EGFR的激活和表達
本研究發現在BMDM和RAW264.7兩種巨噬細胞中證實LPS均可激活EGFR的細胞表面的表達(圖1A-1F)。在CLP(盲腸結扎穿刺)誘導的膿毒癥小鼠模型和臨床急性膿毒癥患者中,巨噬細胞表面的EGFR的表達量均較正常組明顯升高(圖1G-1K)。綜上所述,LPS誘導膿毒癥巨噬細胞表面EGFR的激活,并促進EGFR的表達。
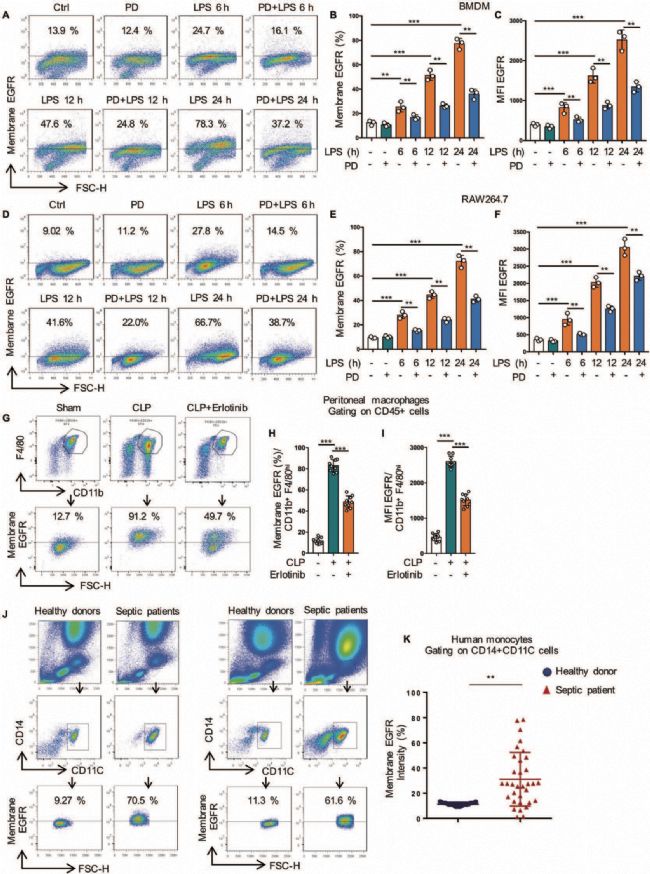
圖1 LPS促進巨噬細胞表面EGFR的表達LPS促進巨噬細胞表面EGFR的表達
2. Rab7a的S72磷酸化促進晚期EGFR內吞
抑制EGFR磷酸化顯著降低了巨噬細胞膜中EGFR的表達,提示EGFR激酶活性在受體運輸中起重要作用。為了發現EGFR激酶新的下游靶點,作者采用定量磷酸化修飾蛋白組學的方法對RAW264.7細胞進行分析(對照組、LPS處理組、EGFR抑制劑(PD168393)+LPS共同處理組)。總共分析了11772個獨特的磷酸位點,對應了8151個肽片段、3041個獨特的蛋白質。磷酸化修飾組學中的KEGG通路分析發現磷酸化肽主要富集于MAPK信號通路、內吞和糖酵解通路(圖2B)。在差異的磷酸化肽段中,只有Rab7a被報道參與了細胞內吞通路,Rab7a GTPase參與調節胞吞介導的蛋白轉運,特別是Rab7A促進了生長因子受體等膜受體從早期核內體到晚期核內體和溶酶體的運輸,因此作者重點研究了RAB7a磷酸化與EGFR轉運之間的關系。修飾組學發現Rab7a在S72位發生磷酸化修飾(圖2C-2D)(經WB和點突變、模擬突變方式證實RAB7a在S72發生了磷酸化修飾(圖3A-3D))。為研究S72磷酸化對Rab7a的GTPase活性的影響,作者分析了LPS處理的巨噬細胞中, Rab7a與CD63(晚期核內體)、LAMP1(自噬體成熟階段相關蛋白)的共定位(圖3E-3J),結果表明Rab7a磷酸化調節了EGFR的后期內吞降解,從而影響了EGFR的膜表達。
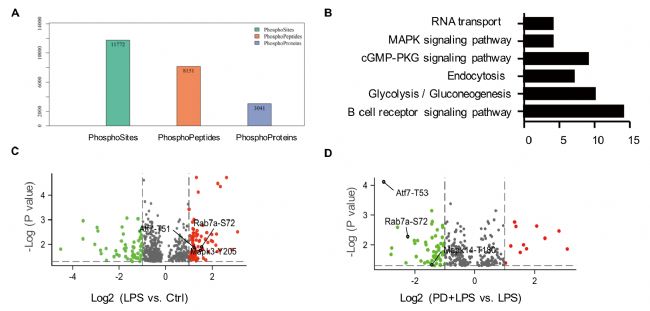
圖2 磷酸化修飾組學KEGG通路和差異表達肽段分析
圖3 Rab7a的S72磷酸化促進晚期EGFR內吞
3. MAPK14磷酸化Rab7a的S72位點
根據磷酸化修飾組學的結果,作者推測Rab7a的S72磷酸化是受EGFR/MAPK14通路影響的。作者通過共定位(圖4A)以及體外、體內(加入MAPK14抑制劑)共沉淀(圖4B-4E)和MAPK14/ Rab7a-S72點突變(圖4G)的方式證實了MAPK14可以直接結合并磷酸化Rab7a,Rab7a S72是MAPK14磷酸化的主要位點。為了了解MAPK14介導的Rab7a磷酸化在EGFR細胞內轉運調控中的作用,作者分析了MAPK14敲除和CD63、LAMP1的共定位(圖4D-4O),結果表明MAPK14介導的Rab7a磷酸化調控了LPS激活的巨噬細胞的晚期內吞和EGFR降解。
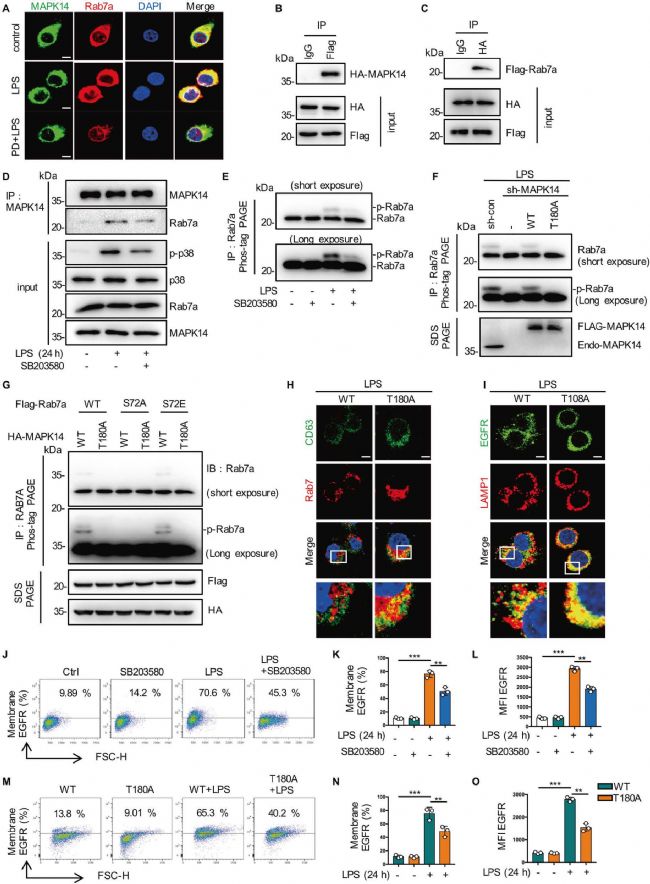
圖4 MAPK14磷酸化Rab7a的S72位點
4. Rab5a介導EGFR在巨噬細胞中的早期內化
生長因子受體的內吞轉運是EGFR信號通路時空調控的重要細胞機制之一。研究發現LPS處理巨噬細胞1小時后,PD168393預處理組中早期核內體(EEA1)和EGFR之間的共定位減弱。由于Rab5a是細胞內吞作用的關鍵調節因子,作者將Rab5a與EGFR進行了免疫共沉淀和免疫熒光染色發現了兩者的共定位。隨后通過Rab5a的敲除以及其效應蛋白抑制劑的添加、Rab5a敲除小鼠的驗證,證實LPS誘導的EGFR早期內化是由Rab5a介導的(圖5)。
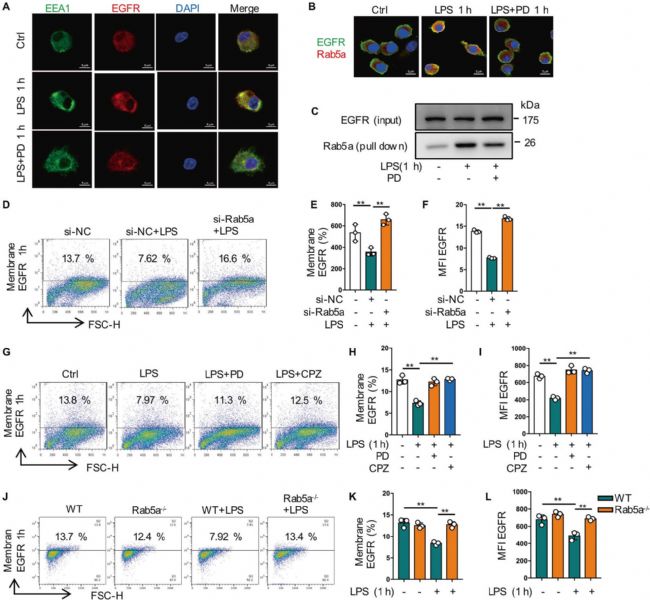
圖5 Rab5a介導EGFR在巨噬細胞中的早期內化
5. Rab10促進EGFR從高爾基體運輸到細胞表面
為了尋找EGFR細胞表面表達的細胞調控機制,作者將重點放在Ras家族的小g蛋白上。Rab10主要參與蛋白質從高爾基體到質膜的運輸。作者發現Rab10定位于高爾基體和EEA1,但不定位于晚期核內體。同時,Rab10和EGFR共定位。此外,Rab10沉默的巨噬細胞表面EGFR表達明顯降低(圖6A-6G)。隨后通過構建Rab10的突變細胞系及EGFR的表面表達,證實Rab10促進EGFR從高爾基體運輸到細胞表面(圖6H-6M)。
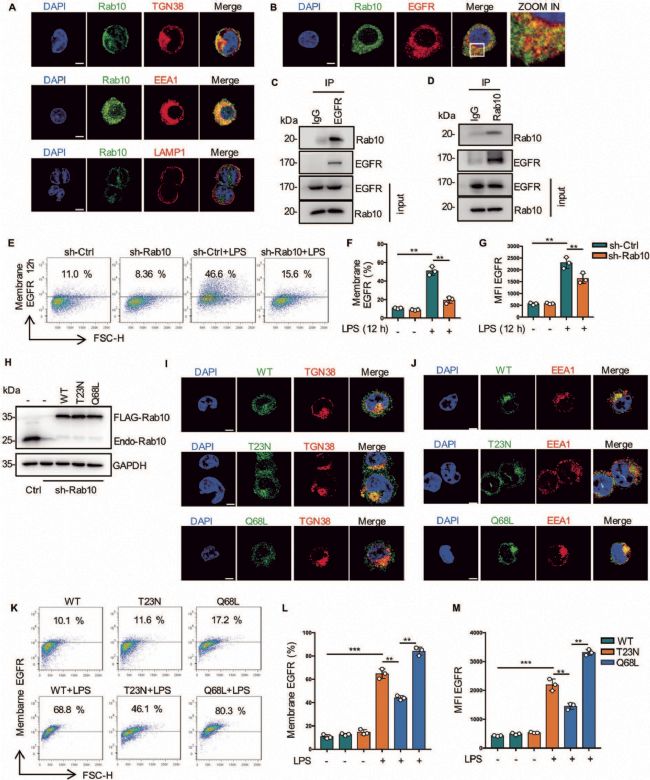
圖6 Rab10促進EGFR從高爾基體運輸到細胞表面
6. 抑制EGFR磷酸化可抑制巨噬細胞中糖酵解依賴性M1極化
為了評估細胞膜EGFR激活對膿毒癥巨噬細胞M1/M2表型平衡的影響,作者使用LPS刺激BMDMs和RAW264.7兩種巨噬細胞向M1表型轉變。研究發現PD168393處理和Erlotinib處理(EGFR抑制劑)均顯著下調了M1標志物iNOS的表達水平,即表明細胞表面的EGFR磷酸化促進內毒素血癥或敗血癥相關的M1巨噬細胞的激活。為了進一步闡明EGFR對內毒素血癥代謝的影響,作者開展了代謝組學分析,結果表明LPS組磷酸戊糖通路(PPP)和糖酵解中間產物增加、PD168393的處理降低HIF-1ɑ和乳酸脫氫酶A (LDHA)蛋白表達。綜上所述,抑制EGFR磷酸化可以減少巨噬細胞對LPS的糖酵解反應(圖7)。
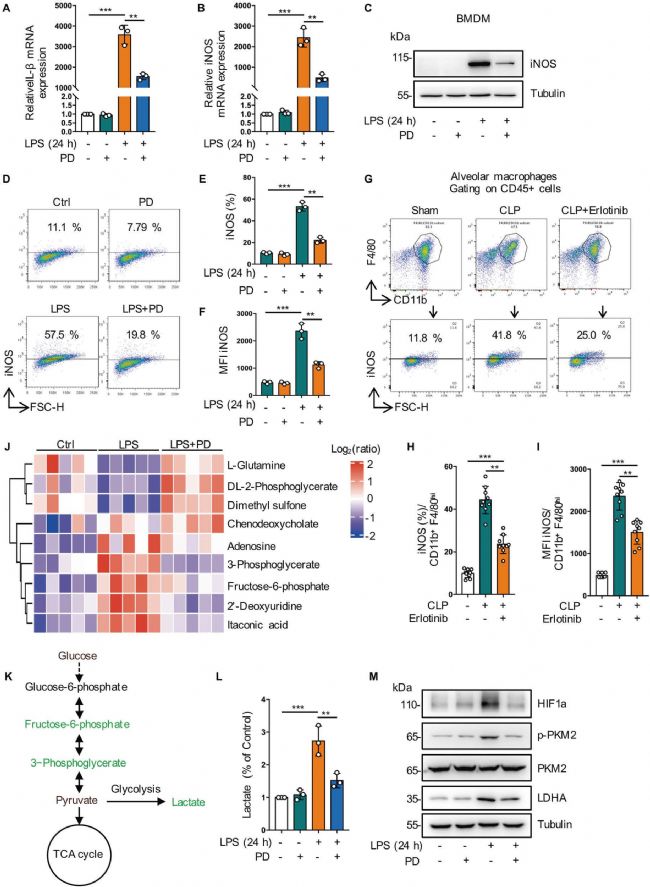
圖7 抑制EGFR磷酸化可抑制巨噬細胞中糖酵解依賴性M1極化
7. 抑制EGFR磷酸化通過激活PPARγ調節谷氨酰胺代謝促進M2極化
作者接進一步研究了EGFR磷酸化在M2表型極化中的作用,結果表明Erlotinib處理后,M2標志物CD206在細胞和小鼠體內均顯著增加,即意味著抑制EGFR磷酸化促進膿毒癥期間M2巨噬細胞極化(圖8A-8J)。PPARγ控制巨噬細胞谷氨酰胺代謝,在M2極化和代謝之間提供聯系,而M2極化需要谷氨酰胺。代謝組學分析表明,PD168393處理后谷氨酰胺水平升高,且PD168393顯著促進了PPARγ的激活,CD206的表達隨著PPARγ的激活而增加(圖8J-8K)。綜上所述,抑制EGFR磷酸化通過激活PPARγ調節谷氨酰胺代謝從而促進M2極化。
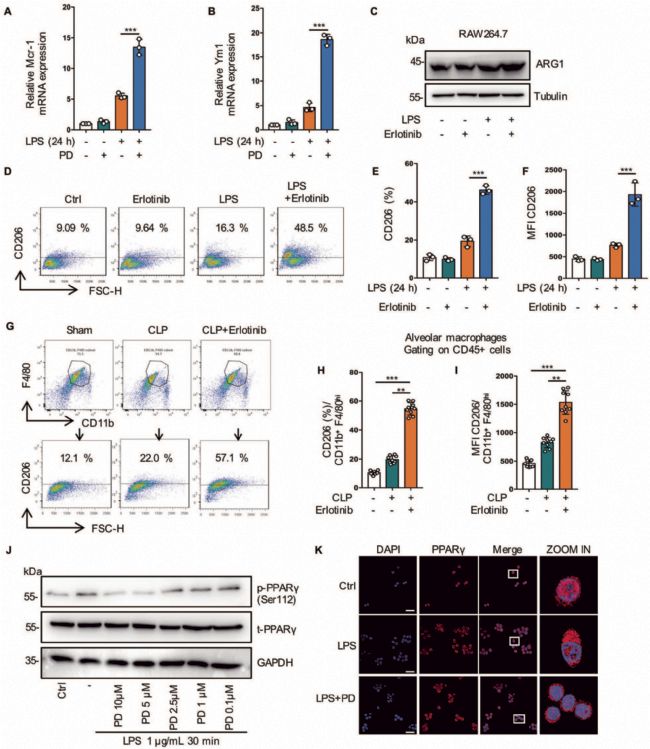
圖8 抑制EGFR磷酸化通過激活PPARγ調節谷氨酰胺代謝促進M2極化
8. 抑制EGFR磷酸化可使M1表型轉變為M2表型,減輕膿毒癥引起的急性肺損傷
為了進一步驗證EGFR抑制劑在體內對炎癥和巨噬細胞極化的影響,應用LPS和CLP誘導的急性肺損傷(ALI)小鼠模型發現,在給予Erlotini的小鼠中,M2標志物CD206的表達增加,而M1標志物iNOS的表達減少。此外,Erlotini明顯減少了中性粒細胞的浸潤減弱了炎癥細胞浸潤、間質水腫和肺泡間隔增厚。綜上表明,EGFR抑制劑可能通過調節體內巨噬細胞極化和減少炎癥來改善膿毒性(圖9)。
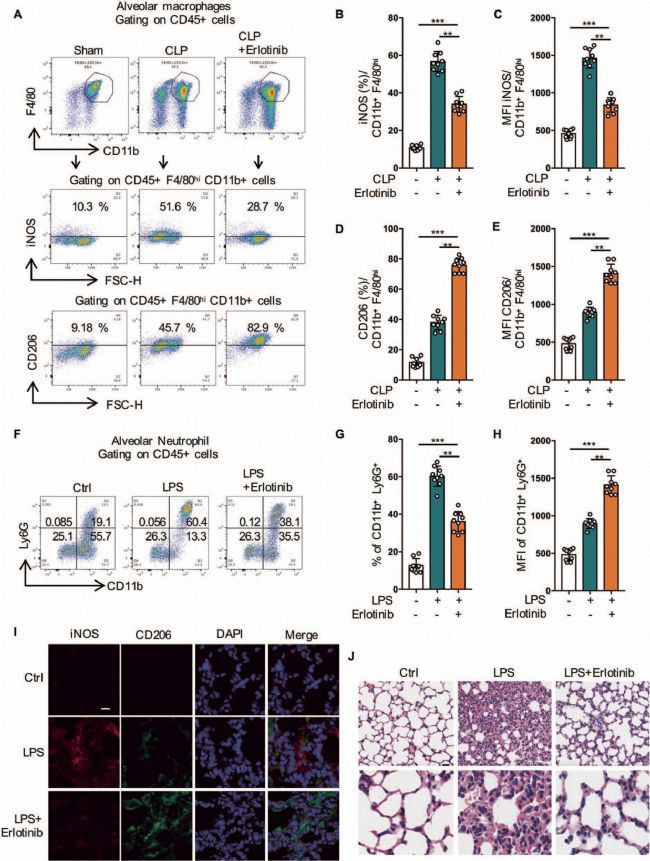
圖9 抑制EGFR磷酸化可使M1表型轉變為M2表型,減輕膿毒癥引起的急性肺損傷
小編小結
該研究發現在LPS作用下,巨噬細胞表面EGFR的表達增強。然后通過磷酸化修飾組和代謝組學等方法揭示了EGFR的整個細胞轉運過程,包括質膜易位、早期內化和晚期內吞,以及調控這些過程的特異性Rab蛋白的分子機制。此外,作者還發現細胞表面EGFR水平調節巨噬細胞M1/M2極化表型轉化,并通過代謝重編程影響膿毒癥誘導的多器官損傷。該研究為EGFR作為膿毒癥治療的潛在靶點提供了有力的證據。



