
PEAKS DB蛋白質質譜數據搜庫功能介紹
自下而上的蛋白質組學(LC-MS/MS)是當今許多蛋白質研究的基礎。對于肽段碎片數據的解析有多種算法。一般的算法通過比較實驗檢測到的碎片離子列表和從蛋白質序列數據庫中獲得的理論碎裂,基于概率的匹配來鑒定肽段。PEAKS DB在進行蛋白鑒定時集成了數據庫搜索和從頭測序。因此,它提供了完整的多肽鑒定,包括各種修飾,突變和未報道過的多肽。
功能特點
- 基于LC-MS/MS數據的搜索引擎
- 未知翻譯后修飾搜索及分析
- 序列突變搜索
- 與從頭測序整合
- 支持CID, HCD, ETD/ECD, EThcD、EAD等多種碎裂模式
靈敏度和準確度
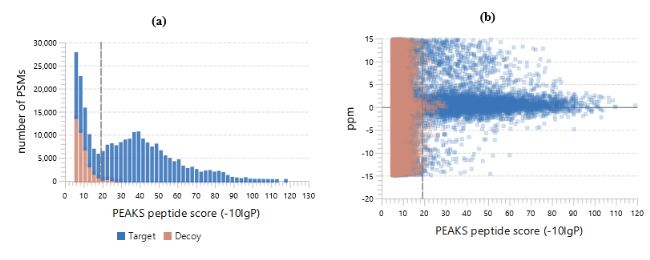
PEAKS DB是基于sequence tag的搜庫算法,保證了鑒定的準確性和靈敏度。獨特的“decoy-fusion”為PEAKS DB提供了非常準確的錯誤發現率(FDR)評估方法。這說明PEAKS DB是一種更加準確、更加靈敏的蛋白鑒定搜索引擎。
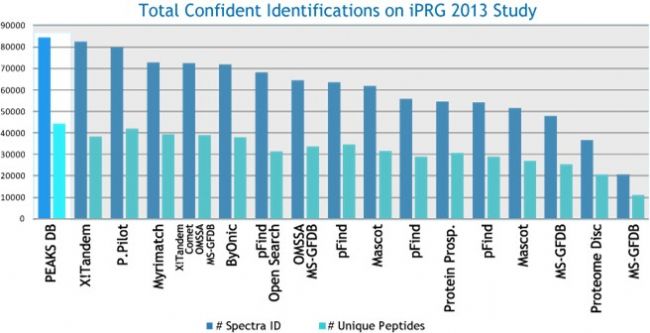
PEAKS DB可以自動地對蛋白鑒定的結果進行統計分析,并在Summary Page中顯示統計數據。用戶可以在工作流的參數設置中通過指定的FDR輕松地過濾數據庫搜索結果。
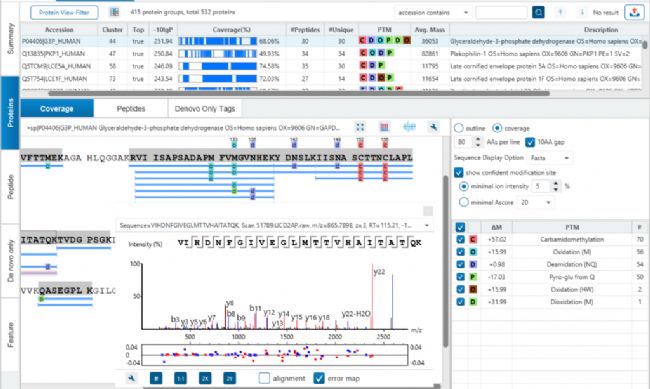
結果可視化
PEAKS 一直以其卓越的結果可視化而聞名。除了以上所述的總覽視圖,用戶還可以從不同角度方便地查看蛋白質鑒定結果。
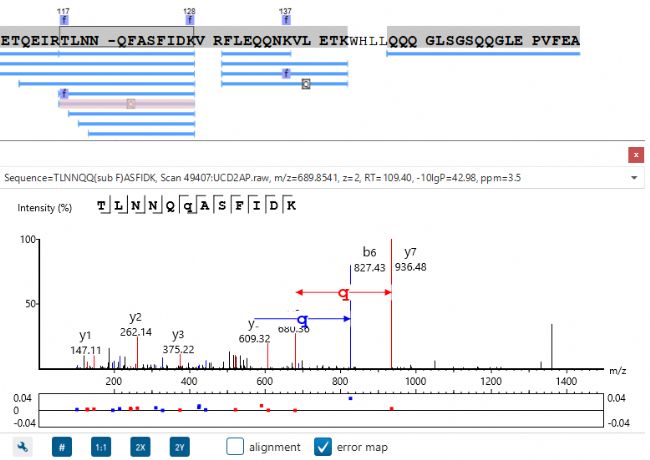
特別是,蛋白質覆蓋視圖(Protein Coverage View)為用戶提供了非常有用的信息,包括在一個蛋白中鑒定到的所有多肽,同時多肽的修飾位點信息和序列突變均會以高亮的字符顯示。
當用戶點擊其中一條鑒定的多肽,PEAKS可以提供肽譜匹配的注釋信息,同時當鼠標在氨基酸殘基上移動時,相應支持的碎片離子信息會在譜圖實時顯示。
特別是,蛋白質覆蓋視圖(Protein Coverage View)為用戶提供了非常有用的信息,包括在一個蛋白中鑒定到的所有多肽,同時多肽的修飾位點信息和序列突變均會以高亮的字符顯示。
當用戶點擊其中一條鑒定的多肽,PEAKS可以提供肽譜匹配的注釋信息,同時當鼠標在氨基酸殘基上移動時,相應支持的碎片離子信息會在譜圖實時顯示。
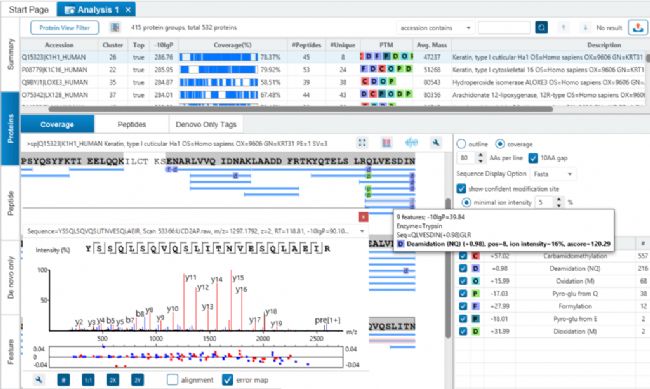
與de novo sequencing結果整合
傳統的搜索引擎的假設是,數據庫中包括了我們樣品中的蛋白質的序列信息,但是,實際上很多我們關心的重要蛋白質,例如單抗,并不包含在任何常用數據庫中。所以這種搜庫策略的缺陷在于,搜庫得到的蛋白最多可以盡可能匹配用于檢索的數據庫,但是當數據庫本身不完整的時候,就會造成大量的多肽不能被鑒定或者鑒定錯誤。
整合了de novo sequencing 的PEAKS DB,能夠提供更為完整的肽段的鑒定,而不依賴于多肽是否存在于數據庫中。
整合了de novo sequencing 的PEAKS DB,能夠提供更為完整的肽段的鑒定,而不依賴于多肽是否存在于數據庫中。
Reference
Tran NH, Qiao R, Xin L, Chen X, Liu C, Zhang X, Shan B, Ghodsi A,
Li M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nature Methods. 16(1), 63-66. 20/12/2018.

Li M. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nature Methods. 16(1), 63-66. 20/12/2018.
如果您想深入了解更多關于PEAKS 蛋白質質譜數據搜庫功能介紹,歡迎掃描下方二維碼關注我們!




Copyright(C) 1998-2025 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com