
PEAKS denovo從頭測序功能介紹--一種不依賴于數據庫的肽段鑒定方法
在串聯質譜中,質譜通過對肽鏈碎裂產生的碎片離子檢測,進而產生ms/ms譜。根據所采用的碎裂方法,可以產生不同的碎片離子類型。未知多肽的測序是在不需要序列數據庫的情況下,從質譜中解析出氨基酸序列。這與另一種流行的肽段鑒定方法--“數據庫搜索”形成了鮮明對比,即在給定的數據庫中搜索與該譜圖正確匹配概率最大(或者錯配概率最小)的肽。當沒有可參考的序列數據庫時,多肽的從頭測序是唯一的選擇。
業界知名的PEAKS系列軟件的核心算法之一就有de novo,可以不依賴于數據庫高效地進行全自動的肽段序列預測。這使得PEAKS軟件成為鑒定未測序生物中新的肽和蛋白質的首選方法。
業界知名的PEAKS系列軟件的核心算法之一就有de novo,可以不依賴于數據庫高效地進行全自動的肽段序列預測。這使得PEAKS軟件成為鑒定未測序生物中新的肽和蛋白質的首選方法。

PEAKS de novo功能特點
高通量、自動化的從頭測序
精確的算法,提供de novo結果可信度打分,確保氨基酸水平的精確度
與database search整合,深度挖掘蛋白質組的未知信息
支持CID、HCD、ETD/ECD 、EThcD、EAD等多種碎裂模式
精確度
PEAKS使用了全面綜合的打分體系,來對于de novo的肽段序列結果的準確性進行打分評估。PEAKS的獨特之處在于local confidence(LC score)評分--評估結果中肽段每個氨基酸分配的可能性。local confidence score將測序的準確度聚焦到氨基酸水平。在圖中, TYEQLAEQNR 是通過了可靠性閾值的序列標簽。

互補的碎片離子
碎片離子對的使用:de novo從頭測序使用由不同的碎裂方式(例如ETD/HCD)產生的成對的譜圖。每個互補的譜圖產生置信度de novo序列標簽用來重構一個肽序列,并對兩個譜圖進行優化。
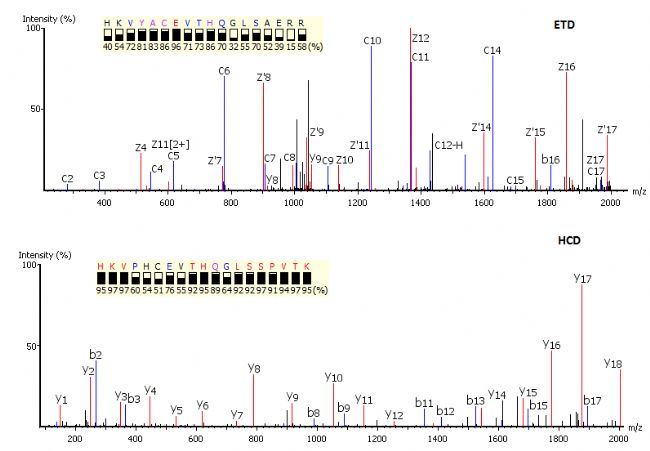
不同譜圖進行測序(優化前)
 互補的譜圖進行測序(優化后)
互補的譜圖進行測序(優化后)
互補的譜圖進行測序(優化后)與Database Search整合
PEAKS的獨特之處在于能夠將多肽de novo測序結果與數據庫搜索結果相結合。多肽de novo序列與蛋白質數據庫條目比對,以提供關于PTM、突變、同源多肽和全新肽段的附加信息。

從多肽從頭測序到蛋白質測序
蛋白質的序列可以由多肽的de novo測序結果中獲得。擁有直接的譜圖碎片離子證據的可靠的de novo多肽序列標簽,可以用來組裝為蛋白質序列。PEAKS de novo應用到蛋白質測序的最佳實例就是在抗體測序方面的軟件PEAKS AB--該軟件可依據質譜二級譜圖所提供的譜峰信息全自動對抗體進行蛋白水平de novo從頭測序及序列拼接的工作,高效自動化地代替人工拼接,從而獲得目標抗體的全序列,為研發工作帶來更大的便利和更好的準確性。

如果您想深入了解更多關于PEAKS denovo從頭測序內容,歡迎掃描下方二維碼關注我們!





Copyright(C) 1998-2025 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com