
PEAKS Q--你應該知道的蛋白定量分析方法
概覽
SILAC 定量
Label-Free 定量
TMT/iTRAQ
為了理解復雜生物系統中單個蛋白質的功能,通常需要測定蛋白質豐度的變化。例如,生物標志物的發現和驗證一般都需要對蛋白質進行定量分析,對其在不同狀態或者處理條件下的表達量變化進行鑒定以及驗證,以此判斷其表達量是否發生顯著性的變化。研究者可以通過PEAKS Q對一組標記或者非標記的LC MS/MS蛋白質樣品的相對變化量進行測定與分析。
功能特點
功能特點
- 基于強度信息的高精度定量
- 對非標記定量和標記定量數據同時適用,包括:SILAC, iTRAQ, TMT(MS2/MS3), N-Terminal
- 支持復雜的實驗設計
- 與數據庫搜索整合
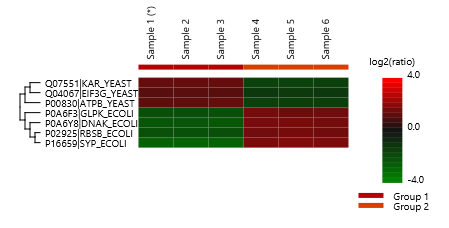
SILAC 定量
細胞培養條件下的氨基酸的穩定同位素標記技術(SILAC) 是鑒定和定量復雜蛋白樣品相對差異變化的一種比較經典的方法。 該技術現在已經成為了體內同位素標記最常用的方法,也是定量蛋白質組學常用的方法。
PEAKS Studio X+ 中的PEAKS Q模塊采用:
01. 改進的SILAC特征峰檢測方法
02. SILAC比對和ID轉移技術成功的提高了定量的準確性和靈敏度。它采用了基于比率的量化方式,這使得super-SILAC實驗的數據分析成為可能。
通過引入多組比較,可以實現時間梯度型的定量。這種新的PEAKS Q模塊能夠實現分別針對單組分析和多組比較分析的配對t檢驗和韋爾奇方差分析。
PEAKS Studio X+ 中的PEAKS Q模塊采用:
01. 改進的SILAC特征峰檢測方法
02. SILAC比對和ID轉移技術成功的提高了定量的準確性和靈敏度。它采用了基于比率的量化方式,這使得super-SILAC實驗的數據分析成為可能。
通過引入多組比較,可以實現時間梯度型的定量。這種新的PEAKS Q模塊能夠實現分別針對單組分析和多組比較分析的配對t檢驗和韋爾奇方差分析。
Label-Free 定量
PEAKS的蛋白相對定量是基于MS1中的離子峰強度信息。在非標記定量(Label-Free)實驗中,樣品通常單獨分開收集進行處理及LC-MS/MS分析。針對實驗結果中的大量數據,PEAKS在離子峰自動對準和比較方面采取了靈敏并精確的算法。通過數據庫搜索得到的MS2中的蛋白質/肽段定性信息也整合進蛋白質的定量分析中。
PEAKS選擇三個豐度最高的獨特肽(Top-three unique peptides)后,排除以下兩種數據,然后對蛋白質調變比率進行計算:
01. 同時具有修飾和未修飾形式的肽
02. 冗余肽段
對于獨立或者相關的樣品在進行分析比較時需要采用的t檢驗或ANOVA分析方法。在進行較大數量的檢驗分析時,對于更準確的錯誤發現率(FDR),P值的解釋也會有所不同。
PEAKS選擇三個豐度最高的獨特肽(Top-three unique peptides)后,排除以下兩種數據,然后對蛋白質調變比率進行計算:
01. 同時具有修飾和未修飾形式的肽
02. 冗余肽段
對于獨立或者相關的樣品在進行分析比較時需要采用的t檢驗或ANOVA分析方法。在進行較大數量的檢驗分析時,對于更準確的錯誤發現率(FDR),P值的解釋也會有所不同。
對于DIA的定量,詳見PEAKS DIA Stramlined工作流的介紹。
TMT/iTRAQ
同位素標簽 (TMT或iTRAQ)具有相同的質量和化學性質,使得輕重同位素可以共洗脫。這些標簽在MS/MS中通過碰撞誘導離解(CID)被從肽段上分離下來,進而用于定量分析。
這個方法有一個挑戰是在質譜鑒定的過程中TMT/iTRAQ標簽的報告基團含量的測定可能會受到其他碎裂片段的干擾。MultiNotch MS3方法通過將多通道和定量靈敏度及精確度結合在一起解決了這個問題。PEAKS同時支持MS2和MS3定量。
有了PEAKS,您現在可以擴大樣本數量進行大規模的蛋白質定量研究,需要參考通道以確保定量的準確性。
這個方法有一個挑戰是在質譜鑒定的過程中TMT/iTRAQ標簽的報告基團含量的測定可能會受到其他碎裂片段的干擾。MultiNotch MS3方法通過將多通道和定量靈敏度及精確度結合在一起解決了這個問題。PEAKS同時支持MS2和MS3定量。
有了PEAKS,您現在可以擴大樣本數量進行大規模的蛋白質定量研究,需要參考通道以確保定量的準確性。
References
-
Yang, W., et al. PEAKS Q: Software for MS-based quantification of stable
isotope labeled peptides. ASMS. WP531. 30/5/2006. -
Xin, L. et al. New Quantitation Software Package Based on PEAKS Protein ID. ASMS. TP 653. 2/6/2008.
-
Chen, C., et al. New Algorithm for Label-Free Protein Quantification.
ASMS. MPB 043. 31/05/2009.
 (點擊圖片即可查看活動詳情)
(點擊圖片即可查看活動詳情)
如果您想深入了解更多關于PEAKS 軟件更多內容,歡迎掃描下方二維碼關注我們!




Copyright(C) 1998-2025 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com