
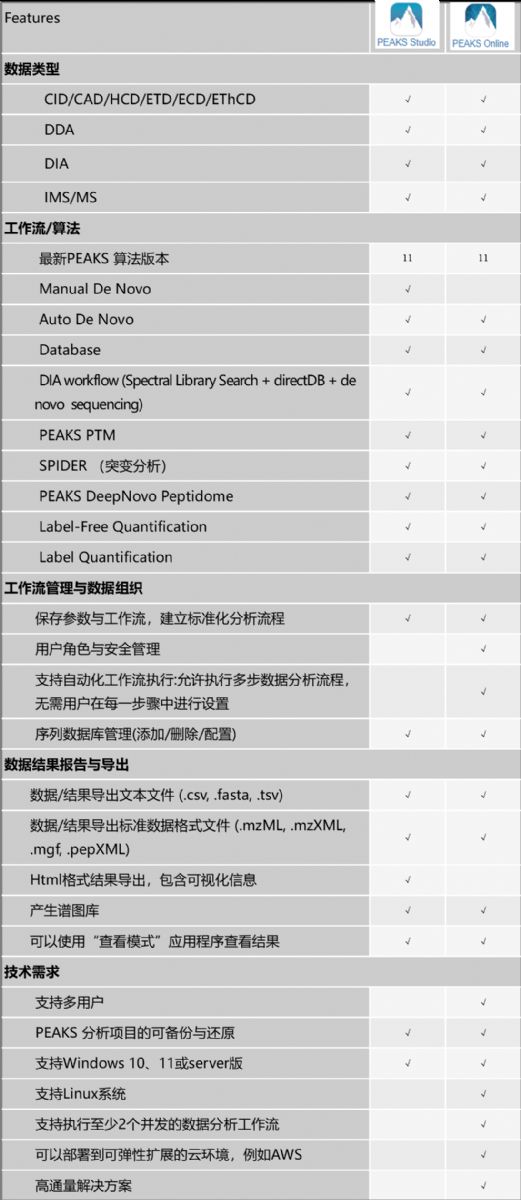
PEAKSĩ°°ŨŲ|―MWĩþ·ÖÎö―âQ·―°ļßxņÖļÄÏ
PEAKSÏĩÁÐÜžþŪaÆ·ĘĮáĶŧųÓÚŌšÏāŲ|ŨV·―·ĻĩÄĩ°°ŨŲ|―MWĩþĩÄÍęÕû―âQ·―°ļĢŽĶÍësĩÄĩ°°ŨŲ|ÓÆ·ÖÐĩÄķāëÄŧōĩ°°ŨŲ|ßMÐÐķĻÐÔšÍķĻÁŋĩÄÏĩ―yÐÔ·ÖÎöĄĢPEAKSÜžþĘĮxÆũSÉĖÖÐÁĒĩÄ·ÖÎöÆ―Å_ĢŽŋÉŌÔÖą―ÓÄŲ|ŨVxžÓÝdÔĘžLC-MS/MSĩþ(oÐčĖáĮ°ÞDQĩþ)ĢŽÔÚPEAKSđĪŨũÁũģĖÖÐßxņßMÐÐëÄšÍĩ°°ŨŲ|čbķĻ(ČįÄî^yÐōĢŽĩþėšÍŨVėËŅËũ)ĢŽ·ŨgšóÐÞïĢĻPTMĢĐ·ÖÎöĄĒÍŧŨĩÄąíÕũŌÔž°ķĻÁŋ·ÖÎöĄĢPEAKSßéĩþŋÉŌŧŊĄĒ―YđûōŨCšÍóļæĖáđĐÁËÔžĄĒŌŨÓÚĘđÓÃĩÄÓÃô―ŧŧĨ―įÃæĄĢ
ÅcÆäËûHŌĀŲĩþėËŅËũĩÄÜžþđĪūßēŧÍŽĢŽPEAKSĘđÓÊĖØĩÄÄî^yÐōÝoÖúÐōÁÐĩþėËŅËũËã·ĻĢŽŨîīóÏÞķČĩØĖáļßëÄķÎĩÄčbķĻЧÂĘĢŽĶÍësĩ°°ŨŲ|―MĖáđĐÁËļüÉîķČĩÄ―âÎöĄĢPEAKS ŧųÓÚÉîķČWÁĩÄžžÐgĢŽÕûšÏÁËķā·NËã·Ļ°üĀĻÄî^yÐōĢĻde novo sequencingĢĐĢŽĩþėËŅËũšÍŨVDėËŅËũĢŽžŊģÉÔÚÓÃôŌŨÓÃĩÄđĪŨũÁũÖÐĢŽÍĻß^ĘđÓÃß@ÐĐ·―·ĻĢŽÄúŋÉŌÔÆÚÍûĖáļßĩ°°Ũ―M/ķāëÄ―MŲ|ŨVĩþĩÄĘī_ÐÔšÍė`ÃôķČĢŽŲxÄÜÎīÖŠÐÞïšÍÍŧŨówĩÄ°lŽFĢŽēĒÄÄî^yÐōĩÄëÄķÎ―YđûÖÐÍÆāÐÂĩÄORFĄĢ
ÅcÆäËûHŌĀŲĩþėËŅËũĩÄÜžþđĪūßēŧÍŽĢŽPEAKSĘđÓÊĖØĩÄÄî^yÐōÝoÖúÐōÁÐĩþėËŅËũËã·ĻĢŽŨîīóÏÞķČĩØĖáļßëÄķÎĩÄčbķĻЧÂĘĢŽĶÍësĩ°°ŨŲ|―MĖáđĐÁËļüÉîķČĩÄ―âÎöĄĢPEAKS ŧųÓÚÉîķČWÁĩÄžžÐgĢŽÕûšÏÁËķā·NËã·Ļ°üĀĻÄî^yÐōĢĻde novo sequencingĢĐĢŽĩþėËŅËũšÍŨVDėËŅËũĢŽžŊģÉÔÚÓÃôŌŨÓÃĩÄđĪŨũÁũÖÐĢŽÍĻß^ĘđÓÃß@ÐĐ·―·ĻĢŽÄúŋÉŌÔÆÚÍûĖáļßĩ°°Ũ―M/ķāëÄ―MŲ|ŨVĩþĩÄĘī_ÐÔšÍė`ÃôķČĢŽŲxÄÜÎīÖŠÐÞïšÍÍŧŨówĩÄ°lŽFĢŽēĒÄÄî^yÐōĩÄëÄķÎ―YđûÖÐÍÆāÐÂĩÄORFĄĢ

PEAKSÜžþĖáđĐķā·NēŋĘð·―°ļĢŽŌÔßmŠļũ·NÓËãhūģĢŽČįŨĀÃæđĪŨũÕūĄĒūÖÓōūWČīî―ĻĩÄ·þÕÆũšÍÔÆhūģĄĢ
ŌŧĄĒxÆũSÉĖÖÐÁĒ
PEAKSÖą―ÓÖ§ģÖíŨÔļũīóÖũÁũŲ|ŨVxÆũSÉĖĩÄËųÓÐÔĘžÎÄžþļņĘ―ĢŽoÐčČΚÎÞDQĄĢĘđÓÃÔĘžĩþĩÄÝÔÚÓÚĢŽÍĻß^Ēß@ÐĐÔĘžĩþĩÄĩŨÓĩþėĮķČëĩ―PEAKSËã·ĻÖÐĢŽĩþÖÐģĘŽFĩÄÐÅÏĒŋÉŌÔß_ĩ―ŨîīóŧŊĄĢoÕÄúÔÚōĘŌÖÐĘđÓÃÄÄ·NxÆũĢŽPEAKSķžáĶēŧÍŽîÐÍĩÄxÆũßMÐÐÁËÏāŠĩÄËã·ĻÓūĢŽÖžÔÚī_ąĢÄú·ÖÎö―YđûĩÄŨîžŅĘī_ÐÔšÍė`ÃôķČĄĢ
ķþĄĒžæČÝDDAšÍDIAĩÄŌŧówŧŊ―âQ·―°ļ
DDAšÍDIAžžÐgķžÔÚŅļËŲ°lÕđĢŽŅÐūŋČËTÐčŌŠŌŧ·N―YšÏÉ·NēÉžŊ·―·ĻücĩÄ·ÖÎö·―·ĻĄĢÔÚđĪI―įĢŽDDAČÔČŧĘĮģĢÓÃĩÄ·―·ĻĢŽß@·N·―·ĻÏāĶŌŅ―ÍęÉÆĢŽĶÓÚÉÐÎīąŧšÜšÃĩØąíÕũĩÄÓÆ·ÓČÆäÖØŌŠĄĢ―üÄęíĢŽDIAÔ―íÔ―ĘÜgÓĢŽŌōéËüūßÓÐÔÚßxķĻĩÄm/z·ķúČēĒÐÐĩØŦ@ČĄËųÓÐĩÄÄļëxŨÓĩÄËųÓÐÆŽķÎëxŨÓĩÄĖØÐÔĄĢß@ŋË·þÁËDDAÖÐßBĀmMS/MSēÉžŊĩÄūÖÏÞÐÔĄĢ
ČŦÐÂDIAđĪŨũÁũ
PEAKS®️éDIAĩþ·ÖÎöĖáđĐ·―ĄĩÄ―âQ·―°ļĄĢÔÚķāëÄĩÄčbķĻÖÐĢŽ―YšÏÁËČý·Nĩþ·ÖÎö·―·ĻĢšŨVDėËŅËũĢŽÖą―ÓÐōÁÐĩþėËŅËũĢĻdirect database searchĢĐĢŽšÍÄî^yÐōĄĢß@ÓĩÄËŅËũ·―·ĻĢŽÆäËŅËũŋÕégÖðu·ÅīóĄĢĘŨÏČĢŽŋÉŌÔÍĻß^ÓÉÏČĮ°ĩÄŨVDčbķĻ―YđûŪaÉúĩÄŨVDėßMÐÐŨVDėËŅËũĢŽÍĻß^ÁËFDRÔOķĻéÖĩß^VšóĩÄķāëÄąŧąĢÁôÏÂíĢŽ]ÓÐÍĻß^ŨVDėËŅËũFDRéÖĩĩÄŋÉŌÔĀ^ĀmßMÐÐÖą―ÓÐōÁÐĩþėËŅËũĄĢÆäÖÐŋÉÐÅĩÄĩþėÆĨÅä―YđûĖížÓĩ――YđûÖÐĄĢČŧšóĘđÓÃÍŽÓĩÄFDR·―·ĻĢŽÔÚĩþėËŅËũÖÐÎīÆĨÅäĩÄŨVDßMŌŧē―ĖÐÐÄî^yÐōĄĢ
ČŦÐÂDIAđĪŨũÁũ
PEAKS®️éDIAĩþ·ÖÎöĖáđĐ·―ĄĩÄ―âQ·―°ļĄĢÔÚķāëÄĩÄčbķĻÖÐĢŽ―YšÏÁËČý·Nĩþ·ÖÎö·―·ĻĢšŨVDėËŅËũĢŽÖą―ÓÐōÁÐĩþėËŅËũĢĻdirect database searchĢĐĢŽšÍÄî^yÐōĄĢß@ÓĩÄËŅËũ·―·ĻĢŽÆäËŅËũŋÕégÖðu·ÅīóĄĢĘŨÏČĢŽŋÉŌÔÍĻß^ÓÉÏČĮ°ĩÄŨVDčbķĻ―YđûŪaÉúĩÄŨVDėßMÐÐŨVDėËŅËũĢŽÍĻß^ÁËFDRÔOķĻéÖĩß^VšóĩÄķāëÄąŧąĢÁôÏÂíĢŽ]ÓÐÍĻß^ŨVDėËŅËũFDRéÖĩĩÄŋÉŌÔĀ^ĀmßMÐÐÖą―ÓÐōÁÐĩþėËŅËũĄĢÆäÖÐŋÉÐÅĩÄĩþėÆĨÅä―YđûĖížÓĩ――YđûÖÐĄĢČŧšóĘđÓÃÍŽÓĩÄFDR·―·ĻĢŽÔÚĩþėËŅËũÖÐÎīÆĨÅäĩÄŨVDßMŌŧē―ĖÐÐÄî^yÐōĄĢ
ČýĄĒSpectral Library Support
PEAKS 11 éÓÃôĖáđĐŨVDėËŅËũĩÄčbķĻđĪŨũÁũĄĢÍĻß^ÉîķČčbķĻĩÄŨVDėĢŽÓÃôŋÉŌÔÝpËÉŋėËŲĩØÔuđĀËûĩÄĩþĄĢ
ŨVDėēéŋīÆũ
ÔÚPEAKS 11ÖÐĢŽÓÃôŋÉŌÔÍĻß^PEAKSŨVDėēéŋīÆũíÖąÓ^ĩØÁË―âËûŨVDėĩÄžđĢŽąãÓÚÍĻß^ĩþĩÄ―yÓÐÅÏĒíÔuđĀŨVDėĩÄŲ|ÁŋĄĢß@ĘđĩÃŋÆWžŌŋÉŌÔÝpËÉĩØŨÔķĻÁxĄĒzēéšÍōŨCŨVDėĢŽŌÔĖáļßčbķĻĄĢÍĻß^ÖąÓ^ĩÄÓÃô―įÃæĢŽÔĘÔSÄúÉÏũšÍēéŋīÓÉPEAKS StudiošÍPEAKS OnlineÉúģÉĩÄÎÄąūļņĘ―PEAKSŨVDėĄĢ
ŨĒŌâĢšÔđĶÄÜHÔÚPEAKS StudioÖÐÖ§ģÖĢŽPEAKS OnlineĩÄÓÃôŋÉŌÔÃâŲMĘđÓÃPEAKS StudioĩÄviewÄĢĘ―íēéŋīĄĢ
ŨVDėēéŋīÆũ
ÔÚPEAKS 11ÖÐĢŽÓÃôŋÉŌÔÍĻß^PEAKSŨVDėēéŋīÆũíÖąÓ^ĩØÁË―âËûŨVDėĩÄžđĢŽąãÓÚÍĻß^ĩþĩÄ―yÓÐÅÏĒíÔuđĀŨVDėĩÄŲ|ÁŋĄĢß@ĘđĩÃŋÆWžŌŋÉŌÔÝpËÉĩØŨÔķĻÁxĄĒzēéšÍōŨCŨVDėĢŽŌÔĖáļßčbķĻĄĢÍĻß^ÖąÓ^ĩÄÓÃô―įÃæĢŽÔĘÔSÄúÉÏũšÍēéŋīÓÉPEAKS StudiošÍPEAKS OnlineÉúģÉĩÄÎÄąūļņĘ―PEAKSŨVDėĄĢ
ŨĒŌâĢšÔđĶÄÜHÔÚPEAKS StudioÖÐÖ§ģÖĢŽPEAKS OnlineĩÄÓÃôŋÉŌÔÃâŲMĘđÓÃPEAKS StudioĩÄviewÄĢĘ―íēéŋīĄĢ
ËÄĄĒÓŅšÃĩÄÓÃô―ŧŧĨ
PEAKSŌÔÆäŨŋÔ―ĩÄ―YđûŋÉŌŧŊķøÂÃûĄĢÓÃôÔÚ―YđûÖÐŋÉŌÔÔLŨēŧÍŽĩÄŌDĢŽĀýČįĢŽĩ°°ŨŲ|ŌDĢŽķāëÄŌDĢŽÉõÖÁÔÚ°ąŧųËáËŪÆ―zēéĄĢÔÚPEAKSĩÄĩ°°ŨŲ|ļēÉwŌDÖÐĢŽÓÃôŋÉŌÔÕŌĩ―ļÐÅdČĪĩÄëÄĢŽēĒÕ{ģöÏāęPĩÄŨVDĄĢžīĘđĘĮPEAKSĩÄģõWÕßŌēŋÉŌÔÍĻß^ÓŅšÃĩÄđĪŨũÁũÔOÖÃÝpËÉĩØ·ÖÎöĩþĢŽČÖÃĩÄ―YđûōŨCŋÉŌÔī_ąĢ―YđûŲ|ÁŋēĒéÓÃôĖáđĐļüķāŋÉÐÅķČĩÄÐÅÏĒĄĢPEAKSDÐÎÓÃô―įÃæÔOÓÖũŌŠÄŌŨÓÚ―âáĢŽ―YđûŋÉŌŧŊšÍōŨCĩÄ―ĮķČíÔOÓĩÄĢŽŌÔÍÖúŅÐūŋČËTÄÖÐŦ@ČĄ·ÖÎöíÄŋĩÄČŦūÖÐÅÏĒĄĢ
ÎåĄĒDe novo Sequencing
PEAKSŌŅģÉéŨÔÓŧŊÄî^yÐōĩÄÐÐIËĘĢŽēĒŌÔÆäĘī_ÐÔĄĒËŲķČšÍŌŨÓÃÐÔķøÂÃûĄĢPEAKSēÉÓÃČŦÃæĩÄÔu·ÖÏĩ―yĢŽĖáđĐūŦī_ĩÄÄî^yÐōĩÄëÄķÎ―YđûĄĢPEAKSĩÄŠĖØÖŪĖÔÚÓÚLocal Confidence (LC)Ôu·ÖĢŽÆäķĻÁxé―MģÉÔķāëÄÖÐĩÄÃŋ°ąŧųËáÔÚÔÎŧÖ÷ÖÅäĩÄŋÉÄÜÐÔĄĢ
ÁųĄĒPEAKS DB: Database Searching
PEAKSÍĻß^ĒÄî^yÐōĩÃĩ―ĩÄķāëÄÐōÁÐÅcÏāŠĩÄĩþėËŅËũĩÄëÄŨVÆĨÅä―YđûÏā―YšÏĢŽéÆäĩþėËŅËũđĪŨũÁũĖáđĐÁËŌŧ·NŠĖØĩÄ·―·ĻĄĢÄî^yÐōĩÄķāëÄÐōÁÐÅcĩ°°ŨŲ|ĩþėlÄŋßMÐÐąČĶĢŽÄÜōéÓÃôĖáđĐęPÓÚPTMsĄĒÍŧŨĄĒÍŽÔīëÄšÍČŦÐÂëÄķÎĩÄî~ÍâÐÅÏĒĄĢPEAKS DBĘđÓÃŧųÓÚÐōÁÐËšĩÄËŅËũËã·ĻĢŽąĢŨCÁËËŅËũĩÄĘī_ÐÔšÍė`ÃôķČĄĢĘđÓÃPEAKS DBĢŽŅÐūŋČËTŋÉŌÔÍĻß^·ÖÎöĩþėÖÐŧōēŧÔÚĩþėÖÐĩÄëÄíÉîķČąíÕũËûĩÄÓÆ·ĄĢ
ÔÚPEAKS DDAđĪŨũÁũģĖÖÐĢŽÍĻß^ÉîķČWÁžžÐgīóīóĖáļßÁËÐÔÄÜĢŽŨîīóÏÞķČĩØĖáļßPEAKS DBËŅËũ―YđûÖÐĩÄķāëÄčbķĻЧÂĘĄĢēÉÓÃÉîķČWÁĩÄËã·ĻíîAyąĢÁôrégĄĒËéÆŽëxŨÓķČĄĒŲ|šÉąČšÍëxŨÓßwŌÆÂĘĢŽĖáļßÁËčbķĻĩÄĘī_ÐÔšÍė`ÃôķČĄĢ
ÔÚPEAKS DDAđĪŨũÁũģĖÖÐĢŽÍĻß^ÉîķČWÁžžÐgīóīóĖáļßÁËÐÔÄÜĢŽŨîīóÏÞķČĩØĖáļßPEAKS DBËŅËũ―YđûÖÐĩÄķāëÄčbķĻЧÂĘĄĢēÉÓÃÉîķČWÁĩÄËã·ĻíîAyąĢÁôrégĄĒËéÆŽëxŨÓķČĄĒŲ|šÉąČšÍëxŨÓßwŌÆÂĘĢŽĖáļßÁËčbķĻĩÄĘī_ÐÔšÍė`ÃôķČĄĢ
ÆߥĒPEAKS PTM-·ŨgšóÐÞïčbķĻÅcķĻÁŋ
ÍĻß^ÕûšÏPEAKS DBšÍde novoyÐō―YđûŽFĶPTMĩÄÉîķČčbķĻĄĢPEAKSîIÏČĩÄËã·ĻŨîīóÏÞķČĩØĖáļßÁËPTMčbķĻšÍPTM profilingĄĢģĢŌĩÄPTMŋÉŌÔÔÚČÎŌâĩÄķĻÐÔ·ÖÎö(Äî^yÐōĄĒPEAKS DBĄĒŨVėËŅËũĄĒPEAKS PTMšÍSPIDER)ÖÐķĻÁxĢŽČŧķøĢŽÖŧÓÐÔÚPEAKS PTMËŅËũÖÐĢŽÓÃôēÅÄÜÍĻß^ÖļķĻŌŧ―MļÐÅdČĪĩÄPTMÁÐąíŧōÄUnimodĩþėÖÐËŅËũËųÓÐ313·NĖėČŧ°lÉúĩÄÉúÎïÐÞïíUÕđPTM·ÖÎöĢŽŌÔ°lŽFĩþÖÐČΚÎîAÆÚŌÔÍâĩÄPTMĄĢPEAKS PTMÍĻß^žŊģÉīóĩÄÄî^yÐōËã·ĻšÍĩþėËŅËũĢŽĢéTÓÃÓÚ°lŽFÔÚ°lÉúĩÄÐÞïĢŽēĒĮŌēŧþß^ÓÚĘÜĩ―ÓËãŲYÔīĩÄÏÞÖÆĄĢĘđÓÃPEAKSŋÉŌÔŨîīóÏÞķČĩØĖáļßčbķĻЧÂĘĢŽēĒØĩŨąíÕũÍësĩ°°ŨŲ|―MÖÐĩÄ·ŨgšóÐÞïĄĢ
°ËĄĒSPIDER: ÐōÁÐŨŪ·ÖÎö
PEAKSÜžþÖÐĩÄSPIDERĢŽĘĮŌŧ·NĢéTÓÃÓÚzyķāëÄÍŧŨēĒßMÐÐŋįÎï·NÍŽÔīÐÔËŅËũĩÄËã·ĻĄĢSPIDERËã·ĻLÔĒde novoÐōÁÐËšÅcĩþėĩ°°ŨŲ|ÆĨÅäĄĢŪ°lŽFï@ÖøĩÄÏāËÆÐÔrĢŽËã·ĻLÔĘđÓÃÄî^yÐōåeÕ`šÍÍŽÔīëÄÍŧŨí―âáēîŪĄĢSPIDERÖžÔÚÖØ―ĻŌŧ“Õæ”ÐōÁÐĢŽŌÔŨîÐĄŧŊÕæÐōÁÐÅcde novoÐōÁÐÖŪégĩÄÄî^Õ`ēîÖŪšÍĢŽŌÔž°ÕæÐōÁÐÅcÆäÖŪégĩÄÍŽÔīëÄÍŧŨĄĢŋÉŌÔÍĻß^PEAKSŌÔÄî^yÐōéŧųĩAĩÄÍŽÔīËŅËũĢŽíŦ@ĩÃĩþėÖÐŌÔÍâĩÄŋÉŋŋÆĨÅäĄĢ
ūÅĄĒPEAKS DeepNovo ķāëÄ―MđĪŨũÁũ
ß@ĘĮŌŧĢéTéķāëÄ―MWĩþ·ÖÎöĩÄŠÓÃöū°ķøé_°lĩÄČŦÐÂĢŲđĪŨũÁũĢŽ―YšÏÁËĩþėËŅËũĄĒÄî^yÐōšÍķāëÄÍŧŨĩÄčbķĻĄĢĀûÓÃķāëÄ―MWĩþžŊÓūDeepNovoÉîķČWÁÄĢÐÍĢŽŋÉï@ÖøĖáļßķāëÄčbķĻĩÄė`ÃôķČšÍĘī_ÐÔĄĢīËÍâĢŽĒde novo peptide (non – canonical)Åcĩþėpeptide (canonical)Ïā―YšÏĢŽÔÚČŦūÖËŪÆ―ļüĘī_ĩØđĀÓFDRĄĢŨî―KÝģö―YđûÖÐĩÄķāëÄąŧ·ÖîéíŨÔÓÚĩþėÆĨÅäĢŽDeepNovoŧōÍŽÔīķāëÄ(ÍŧŨëÄ)ĢŽŋÉŌÔÖą―ÓÝģöÓÃÓÚ―YšÏÓHšÍÁĶšÍÃâŌßÔÐÔîAyĄĢ
ŨĒŌâĢšąūđĪŨũÁũÄŋĮ°ŠÓÃÔÚPEAKS StudiošÍPEAKS OnlineŪaÆ·ÖÐĄĢ
ĘŪĄĒPEAKS Q (ļ―žÓÄĢK) ķĻÁŋ·ÖÎö
éÁËÁË―âÍësÉúÎïÏĩ―yÖÐÎĩ°°ŨŲ|ĩÄđĶÄÜĢŽÍĻģĢÐčŌŠyķĻĩ°°ŨŲ|ØSķČĩÄŨŧŊĄĢŨũéŌŧŋÉßxĩÄļ―žÓÄĢKĢŽÓÃôŋÉŌÔĘđÓÃPEAKS QĢŽÍĻß^LC-MS/MSËÓŧō·ĮËÓķĻÁŋĩÄ·―·ĻÍŽrī_ķĻŌŧ―MÓÆ·ĩÄÏāĶĩ°°ŨŲ|ØSķČŨŧŊĄĢ
ŨĒŌâĢšąūđĪŨũÁũÄŋĮ°ŠÓÃÔÚPEAKS StudiošÍPEAKS OnlineŪaÆ·ÖÐĄĢ
ĘŪŌŧĄĒPEAKS IMS (ļ―žÓÄĢK)ëxŨÓĖĘķČ
ëxŨÓĖĘķČŲ|ŨVĢĻIMS- MSĢĐÍĻß^ÔöžÓëxŨÓ·ÖëxĩÄĩÚËÄūSķČĢŽéÍësĩÄÉúÎïšÍŧŊWŧėšÏÎïĖáđĐÁËÁîČËēÄŋĩÄ·ÖÎöÁũģĖĄĢëxŨÓÍĻß^ū_ârĢŽļųþÆäßwŌÆÂĘßMÐзÖëxĢŽĘđÓÃIMS-MSĢŽŋÉļųþÆäīóÐĄĄĒÐÎ îĄĒëšÉšÍŲ|ÁŋßwŌÆÂĘ ^·ÖëxŨÓĄĢŌōīËĢŽÔÚũ―yŲ|ŨVÖÐŋÉÄÜo·Ļ ^·ÖĩÄëxŨÓĢŽÓÐŋÉÄÜÍĻß^ß@·N·―Ę― ^·Öé_íĄĢ
PEAKS IMSÄĢKĢŽÕûšÏÔÚPEAKSČÎŌâđĪŨũÁũÖÐĄĢŌŨÓÚĘđÓÃĩÄPEAKSDÐÎÓÃô―įÃæĒÔĘžĩþ·ÖéIM-MS, IM-MS/MSšÍLC-IM/MSĄĢŅÐūŋČËTŋÉŌÔšÜČÝŌŨĩØēéŋīŧųÓÚëxŨÓßwŌÆÂĘĩÄËÄūSĖØÕũŨV·åĄĢ
ĘŪķþĄĒPEAKS Glycan ĖĮ·ÖÎöÄĢK (ŋÉßxÄĢKĢĐ
PEAKS GlycanĘĮPEAKS StudioÖÐĩÄŋÉßxÄĢKĢŽéÉîķČĖĮĩ°°Ũ―MĩÄ·ÖÎöĖáđĐÁËŠĖØĩÄ―âQ·―°ļĄĢPEAKS GlycanĀûÓÃŧųÓÚÍęÕûĖĮëÄĩÄ·―·ĻĢŽáĶLC-MS/MSĩþ·ÖÎöÓÆ·ÖÐĩÄĖĮĩ°°ŨĄĢPEAKS GlycanÄĢKĖáđĐÁËŌŧÍęÉÆĩÄĩþėËŅËũšÍÐÂé_°lĩÄËã·ĻĢŽŌÔīŲßMN-šÍo -æūÛĖĮĩÄčbķĻšÍąíÕũĄĢGlycan ProfilingĘĮÔÚĩ°°ŨŲ|ĩÄĖØķĻÎŧücßMÐÐĩÄ(ÎŧÖ÷ÖÎö)ĢŽēĒÍĻß^·ĮËÓķĻÁŋąČÝ^ÓÆ·ÖÐĩÄĖĮëÄØSķČĄĢ
°ËĄĒSPIDER: ÐōÁÐŨŪ·ÖÎö
PEAKSÜžþÖÐĩÄSPIDERĢŽĘĮŌŧ·NĢéTÓÃÓÚzyķāëÄÍŧŨēĒßMÐÐŋįÎï·NÍŽÔīÐÔËŅËũĩÄËã·ĻĄĢSPIDERËã·ĻLÔĒde novoÐōÁÐËšÅcĩþėĩ°°ŨŲ|ÆĨÅäĄĢŪ°lŽFï@ÖøĩÄÏāËÆÐÔrĢŽËã·ĻLÔĘđÓÃÄî^yÐōåeÕ`šÍÍŽÔīëÄÍŧŨí―âáēîŪĄĢSPIDERÖžÔÚÖØ―ĻŌŧ“Õæ”ÐōÁÐĢŽŌÔŨîÐĄŧŊÕæÐōÁÐÅcde novoÐōÁÐÖŪégĩÄÄî^Õ`ēîÖŪšÍĢŽŌÔž°ÕæÐōÁÐÅcÆäÖŪégĩÄÍŽÔīëÄÍŧŨĄĢŋÉŌÔÍĻß^PEAKSŌÔÄî^yÐōéŧųĩAĩÄÍŽÔīËŅËũĢŽíŦ@ĩÃĩþėÖÐŌÔÍâĩÄŋÉŋŋÆĨÅäĄĢ
ūÅĄĒPEAKS DeepNovo ķāëÄ―MđĪŨũÁũ
ß@ĘĮŌŧĢéTéķāëÄ―MWĩþ·ÖÎöĩÄŠÓÃöū°ķøé_°lĩÄČŦÐÂĢŲđĪŨũÁũĢŽ―YšÏÁËĩþėËŅËũĄĒÄî^yÐōšÍķāëÄÍŧŨĩÄčbķĻĄĢĀûÓÃķāëÄ―MWĩþžŊÓūDeepNovoÉîķČWÁÄĢÐÍĢŽŋÉï@ÖøĖáļßķāëÄčbķĻĩÄė`ÃôķČšÍĘī_ÐÔĄĢīËÍâĢŽĒde novo peptide (non – canonical)Åcĩþėpeptide (canonical)Ïā―YšÏĢŽÔÚČŦūÖËŪÆ―ļüĘī_ĩØđĀÓFDRĄĢŨî―KÝģö―YđûÖÐĩÄķāëÄąŧ·ÖîéíŨÔÓÚĩþėÆĨÅäĢŽDeepNovoŧōÍŽÔīķāëÄ(ÍŧŨëÄ)ĢŽŋÉŌÔÖą―ÓÝģöÓÃÓÚ―YšÏÓHšÍÁĶšÍÃâŌßÔÐÔîAyĄĢ
ŨĒŌâĢšąūđĪŨũÁũÄŋĮ°ŠÓÃÔÚPEAKS StudiošÍPEAKS OnlineŪaÆ·ÖÐĄĢ
ĘŪĄĒPEAKS Q (ļ―žÓÄĢK) ķĻÁŋ·ÖÎö
éÁËÁË―âÍësÉúÎïÏĩ―yÖÐÎĩ°°ŨŲ|ĩÄđĶÄÜĢŽÍĻģĢÐčŌŠyķĻĩ°°ŨŲ|ØSķČĩÄŨŧŊĄĢŨũéŌŧŋÉßxĩÄļ―žÓÄĢKĢŽÓÃôŋÉŌÔĘđÓÃPEAKS QĢŽÍĻß^LC-MS/MSËÓŧō·ĮËÓķĻÁŋĩÄ·―·ĻÍŽrī_ķĻŌŧ―MÓÆ·ĩÄÏāĶĩ°°ŨŲ|ØSķČŨŧŊĄĢ
ŨĒŌâĢšąūđĪŨũÁũÄŋĮ°ŠÓÃÔÚPEAKS StudiošÍPEAKS OnlineŪaÆ·ÖÐĄĢ
ĘŪŌŧĄĒPEAKS IMS (ļ―žÓÄĢK)ëxŨÓĖĘķČ
ëxŨÓĖĘķČŲ|ŨVĢĻIMS- MSĢĐÍĻß^ÔöžÓëxŨÓ·ÖëxĩÄĩÚËÄūSķČĢŽéÍësĩÄÉúÎïšÍŧŊWŧėšÏÎïĖáđĐÁËÁîČËēÄŋĩÄ·ÖÎöÁũģĖĄĢëxŨÓÍĻß^ū_ârĢŽļųþÆäßwŌÆÂĘßMÐзÖëxĢŽĘđÓÃIMS-MSĢŽŋÉļųþÆäīóÐĄĄĒÐÎ îĄĒëšÉšÍŲ|ÁŋßwŌÆÂĘ ^·ÖëxŨÓĄĢŌōīËĢŽÔÚũ―yŲ|ŨVÖÐŋÉÄÜo·Ļ ^·ÖĩÄëxŨÓĢŽÓÐŋÉÄÜÍĻß^ß@·N·―Ę― ^·Öé_íĄĢ
PEAKS IMSÄĢKĢŽÕûšÏÔÚPEAKSČÎŌâđĪŨũÁũÖÐĄĢŌŨÓÚĘđÓÃĩÄPEAKSDÐÎÓÃô―įÃæĒÔĘžĩþ·ÖéIM-MS, IM-MS/MSšÍLC-IM/MSĄĢŅÐūŋČËTŋÉŌÔšÜČÝŌŨĩØēéŋīŧųÓÚëxŨÓßwŌÆÂĘĩÄËÄūSĖØÕũŨV·åĄĢ
ĘŪķþĄĒPEAKS Glycan ĖĮ·ÖÎöÄĢK (ŋÉßxÄĢKĢĐ
PEAKS GlycanĘĮPEAKS StudioÖÐĩÄŋÉßxÄĢKĢŽéÉîķČĖĮĩ°°Ũ―MĩÄ·ÖÎöĖáđĐÁËŠĖØĩÄ―âQ·―°ļĄĢPEAKS GlycanĀûÓÃŧųÓÚÍęÕûĖĮëÄĩÄ·―·ĻĢŽáĶLC-MS/MSĩþ·ÖÎöÓÆ·ÖÐĩÄĖĮĩ°°ŨĄĢPEAKS GlycanÄĢKĖáđĐÁËŌŧÍęÉÆĩÄĩþėËŅËũšÍÐÂé_°lĩÄËã·ĻĢŽŌÔīŲßMN-šÍo -æūÛĖĮĩÄčbķĻšÍąíÕũĄĢGlycan ProfilingĘĮÔÚĩ°°ŨŲ|ĩÄĖØķĻÎŧücßMÐÐĩÄ(ÎŧÖ÷ÖÎö)ĢŽēĒÍĻß^·ĮËÓķĻÁŋąČÝ^ÓÆ·ÖÐĩÄĖĮëÄØSķČĄĢ

ĢĻücôDÆŽžīŋÉēéŋīŧîÓÔĮéĢĐ
ČįđûÄúÏëÉîČëÁË―âļüķāęPÓÚPEAKS ÜžþļüķāČČÝĢŽgÓßÃčÏ·―ķþūSīaęPŨĒÎŌĢĄ

ËšĢš
ĩ°°ŨŲ|―MW
ķāëÄ―MW

- ĩ°°ŨŲ|―MWŧųĩA ĢšģĢŌĩ°°ŨÃļĩÄ―YĄĒíÔīž°ÐÔŲ|
- ÓÃôÎÄÕÂĢšĘģÆ·žÓđĪĘđ°ëÏÄÖÐķūÐÔÄýžŊËØpÉŲšÍÉúÎïŧîÐÔëÄÔöžÓ
- áĶSARS-CoV-2ŨŪÖęšËŌÂĪĩ°°ŨĩÄÎŋËÂĄŋđówĩÄé_°lÅcąíÕũ
- DeepSearchĄŠĄŠŧųÓÚÉîķČWÁĩÄļßė`ÃôīŪÂŲ|ŨVĩþËŅė·ÖÎöēßÂÔ
- ÓÃôÎÄÕ Ģš čbķĻēŧÍŽ°l―ÍrégĩÄŅōČéé_·Æ ÖÐÔÚĩÄÉúÎïŧîÐÔëÄ
- ß^ģĖ·ÖÎöžžÐgĢĻPATĢĐÖŠŨRđÜĀíÜžþÏĩ―yĩÄ―é―Bž°ŠÓÃ
- ÓÃôÎÄÕÂĢšŧųÓÚLC-MS/MSčbķĻ―zūI·NîŌÔĖáđĐđÅīúĘđÓÃŌ°ÉúÐQ―zĩÄŨCþ
- NatureŨîÐÂŅÐūŋģÉđûĢšČÔīÐÔëÄūSģÖÖÐÐÉņ―Ïĩ―yĩÄÃâŌßŧíÃâCÖÆ
- BSIŅûÄú ĒžÓ2025ÄęĩÚÁųÃČŦøĖĮŋÆWĮāÄęŋÆžžÕŊ
- ÖvŨųĢšMOEÜžþÔÚPROTACŅÐūŋÖÐĩÄĮ°ŅØŠÓÃ
- ÖvŨųĢšXybion PristimaÓÎïōđÜĀíšËÐÄIÕÄĢK
- ÖvŨųĢšÐÂŌŧīúŧŊW―YūÝÆũMarvinđĶÄÜž°ÆäŠÓÃ―é―B
- DeepProteomicsÔÆÖvĖÃĢšŌŧŨũ°éÄãÐÐÏĩÁÐÖvŨų
- ÅRīēĮ°ōĘŌĩÄĩŨÖŧŊŨļï--Xybion Pristima·―°ļ
- 2025ĩÚĘŪÆßÃÖÐøÕûšÏÉúÎïÓąūWīóþÍĻÖŠ
- ŧîÓÕũžŊĢš2025Äęĩ°°ŨŲ|―MWÔÚūÖvŨųÖũî}



Copyright(C) 1998-2025 ÉúÎïÆũēÄūW ëÔĢš021-64166852;13621656896 E-mailĢšinfo@bio-equip.com