
細胞類型注釋不同類別方法及常見的一些軟件或工具介紹
細胞類型注釋是scRNA-seq數據分析中的關鍵一環。傳統的手動注釋方法通常包含兩個步驟:首先使用無監督學習對細胞進行聚類,然后依據聚類結果中典型的差異表達基因來將細胞類群注釋為不同的類型。這一過程十分繁瑣耗時,并且依賴先驗知識,帶有一定的主觀傾向;相比之下,自動注釋工具和方法具有快速簡潔的優點。scRNA-seq細胞的自動注釋一般可概括為三種類型[1],下圖展示了不同類別方法中常見的一些軟件或工具(圖1)。
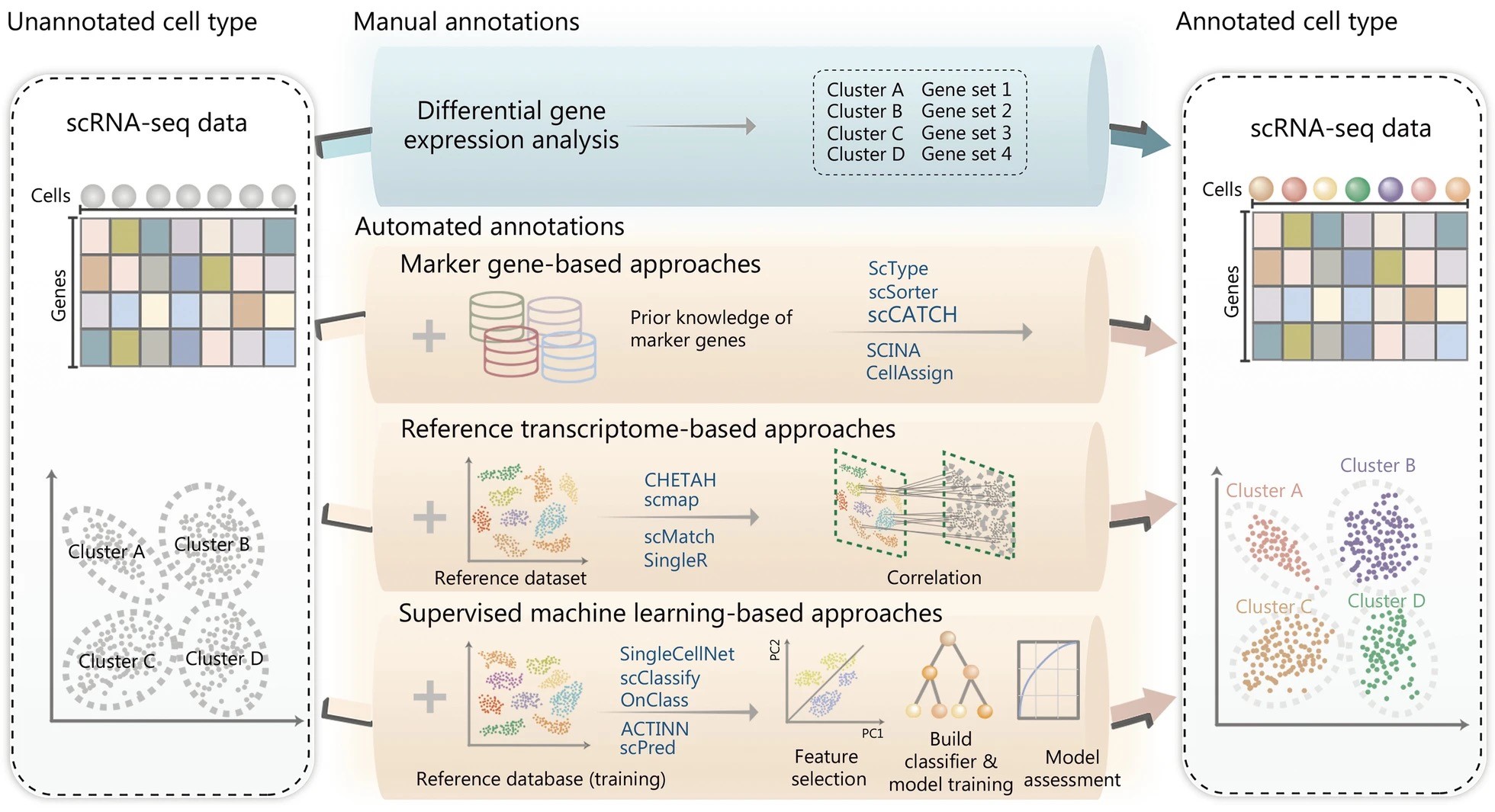
圖1:scRNA-seq細胞注釋的典型策略及代表性方法
第一種方法是基于Marker基因的細胞自動注釋,依據一些特定基因來對細胞進行標記,判定細胞類型,選定的Marker基因或基因集應當具有細胞類型特異性并且能夠穩定表達的。因此,該方法非常依賴Marker的準確性。獲取Marker基因的方法有很多種,通常可以從一些公共數據庫或者文獻中得到。CellMarker[2]和PanglaoDB[3]是較為常見的存儲人類和小鼠組織中多種細胞類型標記物的在線資源。第二種方法是基于參考轉錄組數據集的細胞注釋。使用具有細胞類型標記的數據集作為參考,通過關聯輸入數據的基因表達值,從而將參考數據中的細胞或簇標簽轉移到具有類似基因表達譜的輸入數據中的未標記細胞或簇。該方法較為依賴參考數據的來源和可靠性。最后,第三種方法利用了有監督機器學習,將帶有細胞標記的參考數據作為訓練集,通過機器學習方法得到一個模型(分類器),然后用于預測待注釋細胞的細胞類型。同樣,該方法依賴參考數據的質量。下面,我們從上述方法中各挑選一個工具進行闡述。
01 CellAssign
CellAssign[4]是一種基于概率圖模型的方法,于2019年發表在Nature Methods上。它利用已有的細胞類型Marker基因的先驗知識,通過推斷細胞類型的概率來將未知數據分配到不同的細胞類型(圖2)。具體來說,CellAssign將每個細胞表示為一個隨機變量,該變量表示細胞屬于各細胞類型的概率。同時,它還考慮了批次和樣本效應的影響,通過引入協變量來調整這些效應。CellAssign還考慮了基因的表達變異性,并使用原始計數矩陣和負二項分布來建模。通過最大化后驗概率估計,CellAssign可以推斷每個細胞屬于各個細胞類型的概率,并將細胞分配到具體的細胞類型中。與其他方法相比,CellAssign在準確性和F1得分方面表現更好,并且能夠控制批次效應和樣本效應。
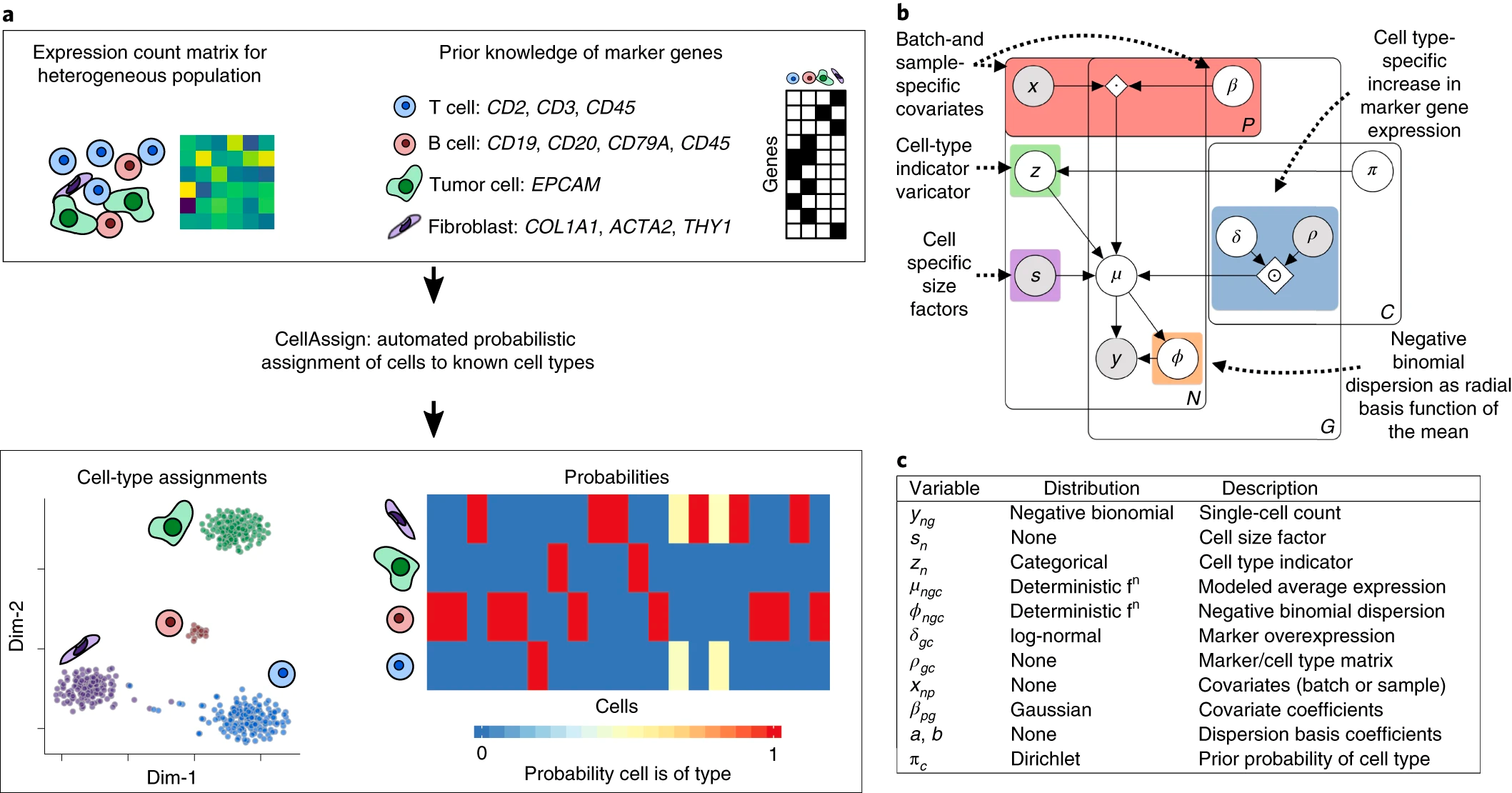
圖 2:CellAssign概覽。(a) 使用CellAssign進行細胞注釋,每個細胞被概率地分配到給定的細胞類型,而不需要任何手動注釋或干預,并且考慮到批次效應的影響。(b) 圖像展示了包含隨機變量、數據、分布假設等在內的細胞類型分配概率模型。(c) 在CellAssign概率模型中使用的隨機變量的描述,以及它們的先驗分布。
◆ 軟件的下載可參考如下方式(任選其一):
# 從GitHub下載
1. install.packages("tensorflow")
2. tensorflow::install_tensorflow(extra_packages='tensorflow-probability')
3. install.packages("devtools")
4. devtools::install_github("Irrationone/cellassign")
# 通過Conda下載
conda install -c conda-forge -c bioconda r-cellassig
◆ 下面以軟件自帶的示例數據進行基礎用法演示:
# 加載R包
1. library(SingleCellExperiment)
2.library(cellassign)
3. library(scran)
# 加載內置數據集
4. data(example_sce)
5. data(example_marker_mat)
# 計算細胞標準化因子(示例數據已經包含該部分信息,如果是新數據,需要重新計算。推薦使用scran包中的computeSumFactors 函數。注意:在計算標準化因子之前最好不要對表達矩陣的基因進行篩選
6. # example_sce <- computeSumFactors(sce)
7. s <- sizeFactors(example_sce)
# 開始運行cellassign。這一步關鍵的地方在于,必須要保證輸入的表達矩陣只包含marker基因的部分
8. fit <- cellassign(exprs_obj =
example_sce[rownames(example_marker_mat),],
marker_gene_info = example_marker_mat,
s = s,
learning_rate = 1e-2,
shrinkage = TRUE,
verbose = FALSE)
02 SingleR
在一篇研究肺纖維化相關巨噬細胞亞型的文章中[5],作者同時開發了一種單細胞轉錄組數據的細胞類型注釋工具——SingleR。它通過將未知類型的單細胞的基因表達與參考數據集中已知細胞類型的基因表達進行相關性分析,來確定每個單細胞的細胞類型。它的注釋過程包括以下幾個步驟:
1、針對每個單細胞獨立進行注釋。首先,計算單細胞表達與參考數據集中每個樣本的Spearman相關系數。這個相關性分析僅針對參考數據集中的變異基因進行;
2、根據參考數據集中的命名注釋,將每個未知細胞的多個相關系數整合,為了防止由于參考樣本的異質性而導致錯誤分類,SingleR使用相關系數的80%作為閾值;
3、SingleR重新運行相關性分析,但僅針對上一步中排名靠前的細胞類型。并且這個分析僅針對囊括的細胞類型之間的變異基因進行。最低相關性值的細胞類型被移除(或者比最高值低0.05以上),然后重復這個步驟,直到只剩下兩個細胞類型為止。
最后一次運行后,對應于最高值的細胞類型被分配給單個細胞(圖3)。
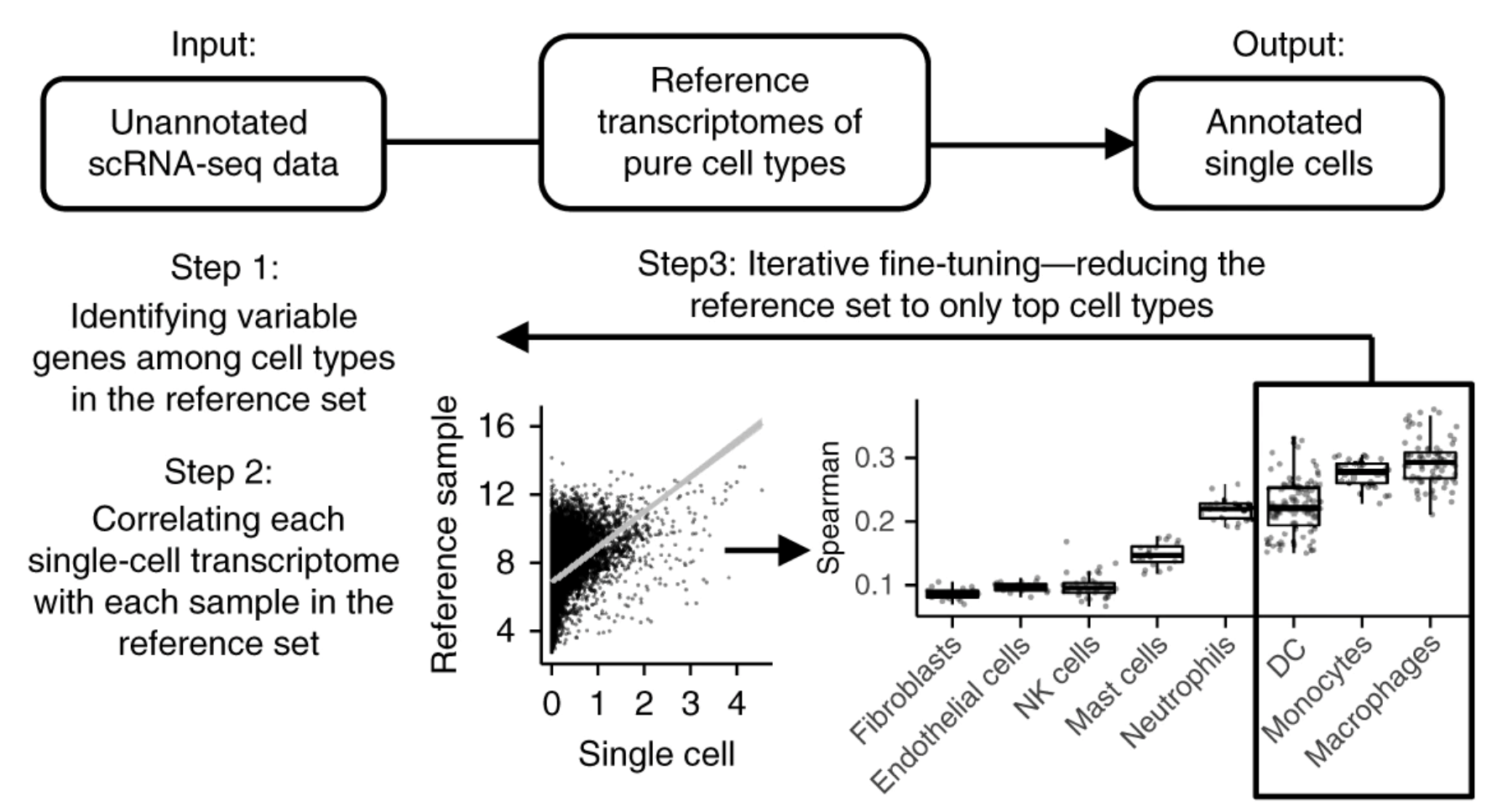
圖 3:SingleR原理示意圖
◆ SingleR包自帶一些參考數據集,詳細信息可通過官方網站查詢,下面對它的基礎用法進行演示:
# 從Bioconductor下載
1. BiocManager::install("SingleR")
# 加載R包及測試數據
2. library(celldex)
3. library(SingleR)
4. library(scRNAseq)
5. hpca.se <- HumanPrimaryCellAtlasData()
6. hESCs <- LaMannoBrainData('human-es')
7. hESCs <- hESCs[,1:100]
# 注釋
8. pred.hesc <- SingleR(test = hESCs, ref = hpca.se, assay.type.test=1, labels = hpca.se$label.main)
03 CellTypist
CellTypist[6]是一個用于自動注釋人類組織中免疫細胞的工具。它的原理是通過整合來自不同組織的細胞數據,并使用機器學習方法訓練模型,實現對細胞類型的準確分類(圖4)。研究人員收集了來自20個不同組織的細胞數據,并對這些數據進行深度篩選和整合,以獲得兩個層次的細胞類型信息。然后,他們使用邏輯回歸和隨機梯度下降學習的方法訓練了模型。模型的性能通過精確度、召回率和全局F1分數進行評估,結果顯示在高層次和低層次的細胞類型分類上都達到了約0.9的準確性。CellTypist能夠識別出不同的細胞亞群,包括T細胞、B細胞、單核巨噬細胞等,此外,它還能夠自動注釋細胞的詳細亞型,提供了對免疫細胞群體的深入了解。它的預測結果對不同數據集之間的差異具有魯棒性,包括基因表達稀疏性和批次效應等。
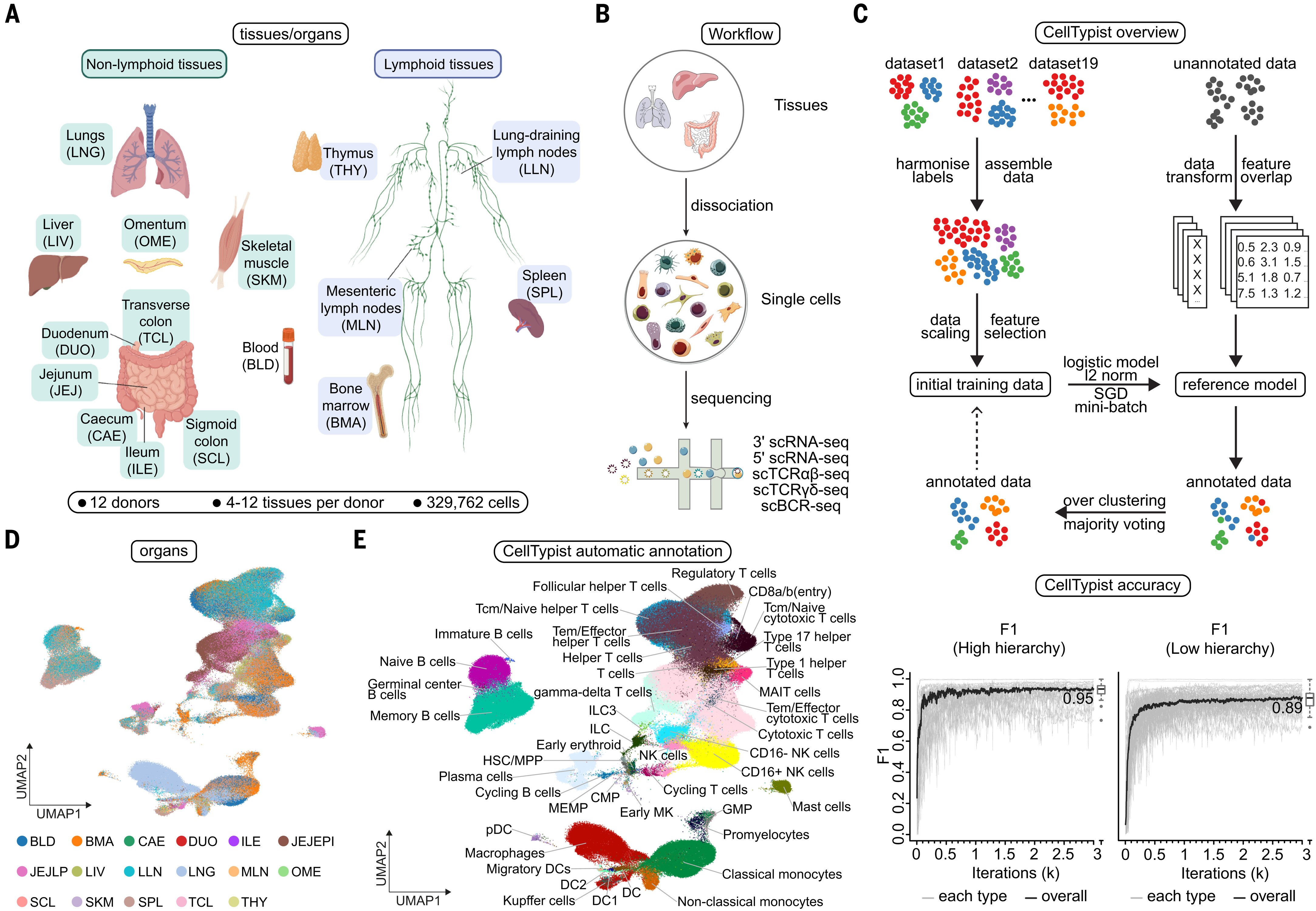
圖 4:CellTypist原理示意圖。CellTypist的工作流程包括數據收集、處理、模型訓練和細胞類型預測
CellTypist既可以作為一個注釋工具使用,也可以作為一個免疫細胞相關的數據庫(圖5)。在其官網上,我們可以通過Encyclopedia查詢特定細胞類型的相關Marker信息,也可以輸入特定基因來查看其在不同組織不同細胞類型中的表達情況(圖6a-c)。
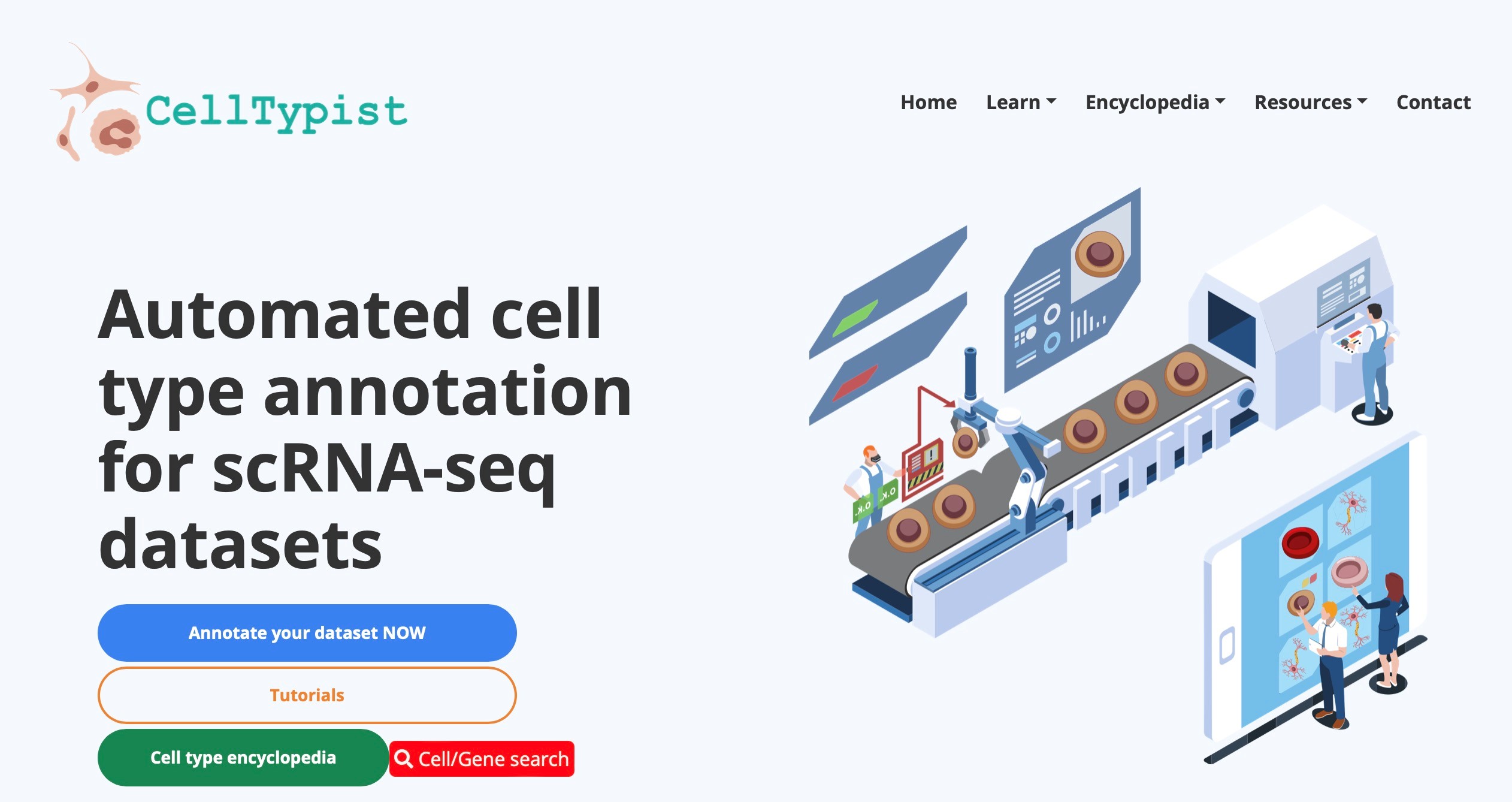
圖 5:CellTypist官方網站示意圖
而作為工具使用時,CellTypist提供了多種運行方法。你可以通過命令行工具進行細胞注釋,可以在Python環境下使用,可以通過Docker運行。此外,你也同樣可以在其官網進行在線分析(圖6d)。下面我們介紹幾種簡單的用法。
◆ 軟件的下載:
# pip方式下載
1. pip install celltypist
# conda方式下載
2. conda install -c bioconda -c conda-forge celltypist
◆ Python中使用:
# 模塊導入
1. import celltypist
2. from celltypist import models
# 下載(初次使用時)并導入模型
3. models.download_models()
4. model = models.Model.load(model = 'Immune_All_Low.pkl')
# 細胞注釋
# 注意1:輸入數據的類型比較寬松,可以是環境內部變量,可以是外部文件;
# 可以是表達矩陣,也可以是AnnData對象;
# 注意2:當輸入表達矩陣時,需要保持行為細胞,列為基因;可以通過添加參數”--transpose-input”轉置
5. predictions = celltypist.annotate(input_file, model = model)
◆ 命令行使用:
# 下載模型(初次使用時)
1. celltypist –update-models
# 細胞注釋
2. celltypist --indata /path/to/input/file --model Immune_All_Low.pkl --outdir /path/to/outdir
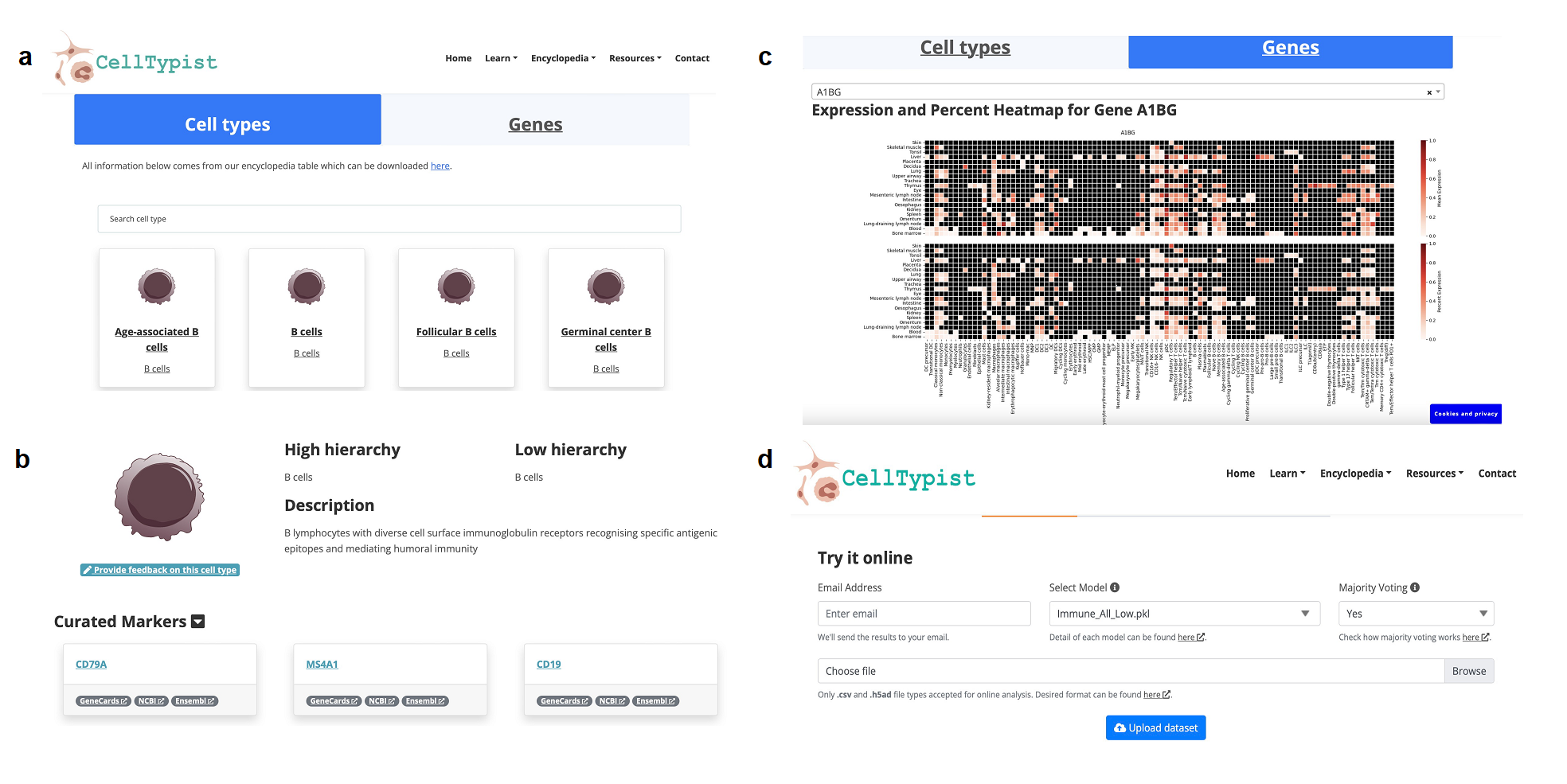
圖 6:CellTypist在線工具。(a) 細胞或基因檢索界面。(b)查詢特定細胞類型后相關信息展示。(c)查詢特定基因后熱圖展示。(d) CellTypist在線注釋工具界面展示。
盡管自動注釋工具可以自動預測scRNA-seq數據的細胞類型,但與傳統的基于Marker的方法一樣,它們仍然需要關于細胞類型的一些先驗知識。前文我們提到了自動注釋工具的三種分類方法,但究其本質,仍然都是從一個reference開始的,無論是Marker還是數據集。因此,盡管使用同一種方法,但如果使用不同的參考數據(訓練模型、細胞特異性Marker等),仍然可能導致截然不同的注釋結果。所以,在初期的準備階段,如何選擇參考數據需要格外謹慎。
另外,依據注釋分辨率水平的不同,這些工具還可以被進一步分成cell-based和cluster-based,即注釋到單個細胞或注釋到簇。研究發現[7],參考數據的組成也會顯著影響自動注釋工具的性能。測試中,注釋結果的準確率會隨著參考數據中每種細胞類型的數量的增加而逐漸提高。當參考數據中包含的某一細胞類型數量較小時,使用注釋單個細胞的方法可能不再合適。
在一些基準測試中,不同的研究給出了不同的結論。比如在Tamim等人的研究中[8],作者使用了27套測試數據,基于準確性、未分類細胞的百分比和計算時間等系統性評估了22種注釋工具或方法。結果表明,支持向量機(SVM)分類器的整體表現最好。在另幾項研究中[7,9],作者都沒有明確表明哪種方法在所有評估指標上表現最好,而是在不同的指標下,不同方法具有不同的表現性能。因此,對于自動注釋來說,更重要的可能不是工具的選擇,而是參考數據的質量以及與待注釋樣本的匹配性。
最后,Zoe A. Clarke等人在21年給出了一份關于scRNA-seq細胞注釋的建議[10]。如圖所示(圖7),對于一個新的數據來說,細胞注釋應當按照如下步驟進行。①自動注釋:建議首先嘗試使用自動注釋方法。自動注釋方法可以快速進行細胞注釋,并且在處理大規模數據集時效率高。自動注釋的結果可能會受到所選擇的參考數據的影響,因此可以嘗試不同的Marker基因組合或數據集來優化注釋結果;②手動注釋:如果自動注釋的結果不夠準確或存在沖突/缺失的細胞標簽,就需要進行手動注釋。手動注釋需要人工檢查每個細胞的特征,并參考各種資源來確定其功能和細胞類型。通常,可以通過可視化每個已知Marker基因在二維數據圖上的表達情況來進行手動注釋。此外,還可以通過查閱文獻和挖掘已有的單細胞轉錄組數據來尋找額外的Marker基因;③最后,細胞注釋的結果可以進行獨立驗證,例如新的驗證實驗,或與互補數據進行比較,例如空間轉錄組學數據。
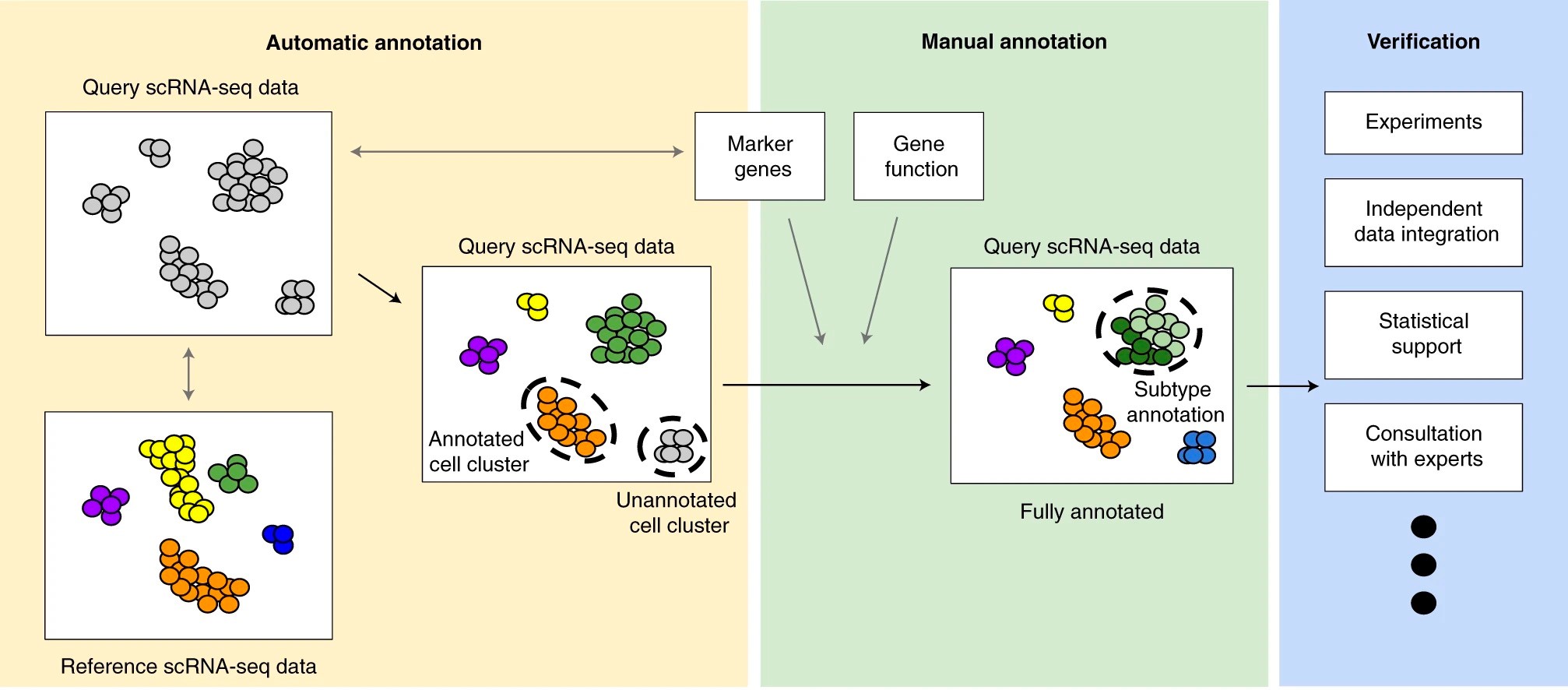
圖 7:細胞注釋過程由三個主要步驟組成:自動細胞注釋、手動注釋和驗證。
參考文獻:
[1] Su, M., Pan, T., Chen, QZ. et al. Data analysis guidelines for single-cell RNA-seq in biomedical studies and clinical applications. Military Med Res 9, 68 (2022).
[2] Hu C, Li T, Xu Y, Zhang X, Li F, Bai J, Chen J, Jiang W, Yang K, Ou Q, Li X, Wang P, Zhang Y. CellMarker 2.0: an updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023 Jan 6;51(D1):D870-D876.
[3] Oscar Franzén, Li-Ming Gan, Johan L M Björkegren, PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data, Database, Volume 2019, 2019, baz046, doi:10.1093/database/baz046
[4] Zhang, A.W., O’Flanagan, C., Chavez, E.A. et al. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat Methods 16, 1007–1015 (2019).
[5] Aran, D., Looney, A.P., Liu, L. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20, 163–172 (2019).
[6] C. Domínguez Conde et al. ,Cross-tissue immune cell analysis reveals tissue-specific features in humans.Science 376,eabl5197(2022).
[7] Huang Q, Liu Y, Du Y, Garmire LX. Evaluation of Cell Type Annotation R Packages on Single-cell RNA-seq Data. Genomics Proteomics Bioinformatics. 2021 Apr;19(2):267-281.
[8] Abdelaal T, Michielsen L, Cats D, Hoogduin D, Mei H, Reinders MJT, Mahfouz A. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 2019 Sep 9;20(1):194.
[9] Xie B, Jiang Q, Mora A, Li X. Automatic cell type identification methods for single-cell RNA sequencing. Comput Struct Biotechnol J. 2021 Oct 20;19:5874-5887.
[10] Clarke, Z.A., Andrews, T.S., Atif, J. et al. Tutorial: guidelines for annotating single-cell transcriptomic maps using automated and manual methods. Nat Protoc 16, 2749–2764 (2021).



