
scMegaÖúÁĻscATAC-seqĩūÅcscRNA-seqĩūĩÄÂēĪˇÖÎö
Îŧ°ûRNAyĐōŧŧĐgŖ¨scRNA-seqŖŠŊŌĘžÁËÎŧ°ûĩÄģųŌōąíß_ĮérŖŦÎŧ°ûATACyĐōŧŧĐgŖ¨scATAC-seqŖŠŖ×ĸĶÚÎŧ°ûĩÄČžÉĢŲ|é_ˇÅĐÔŖŦī@ŦFÁËŧ°ûČĩÄģųŌōÕ{ŋØĮérĄŖɡNŧŧĐgŊYēĪĘšĶÃŋÉŌÔ¸üēÃĩØÍÆāŧ°ûȲŋĩÄģųŌōÕ{ŋØžWŊjĄŖĩĢĘĮß@ĐŠĩūĩġÖÎöŊŗŖŌĒĘšĶÃ˛ģÍŦˇÖÎöš¤žßˇÖeßMĐĐĄŖĀũČįŖŦĻĶÚscRNA-seqĩūŌģ°ãĘšĶÃSeurat°üˇÖÎöŖŦļøscATAC-seqĩūtĘšĶÃArchR°üíßMĐСÖÎöēÍÜÛEÍÆāŖŦĻŪDäŌō×ĶŖ¨transcription factorsŖŦTFsŖŠĩÄģîĐÔšĀytĶÉchromVAR°üßMĐĐŖŦÖTČį´ËîĄŖß@ĘšĩÃĻÎŧ°ûģųŌōÕ{ŋØžWŊjĩġÖÎö×ĩÃĘŽˇÖÍësēͲģąãĄŖ
ģųĶÚß@ĐŠî}ŖŦ scMegaß@ĶŌģÕûēĪÁËļāˇNŦFĶĐĩūˇÖÎöˇŊˇ¨ĩÄļāŊMWˇÖÎöš¤žßĒß\ļøÉúĄŖÔš¤žß°üēŦÁËĩūÕûēĪĄĸŧ°ûÅäĻĄĸÍÆāÎrégÜÛEĄĸTFsēYßxĄĸļ¨ÁŋģųŌōÕ{ŋØžWŊjēÍÔö×ĶTFs-ģųŌōģĨ×÷×ReĄŖ
žßķwļøŅÔŖŦscMegaŋɡÖéČũÖ÷ŌĒ˛ŊķEŖē
ĸŲÎŧ°ûļāŊMWĩūÕûēĪŖŦēōßxTFsēÍģųŌōĩÄ×ReÅcēYßxēÍģųŌōÕ{ŋØžWŊjˇÖÎöĄŖÔÚÎŧ°ûļāŊMWĩūÕûēĪÖĐŖŦĀûĶÃSeuratĩÄĩäĐÍĪāęPˇÖÎöŖ¨CCAŖŠĸscRNA-seqĩūÅcscATAC-seqĩūßMĐĐÕûēĪŖŦČįšû´æÔÚÅú´ÎЧĒŖŦÔŲĀûĶÃHarmonyßMĐĐĐŖÕũŖŦĘšĶÃēķOptMatchĸscRNA-seqÅcscATAC-seqĩÄŧ°ûßMĐĐÆĨÅäŖŦŊ¨ŌģÎļāÄŖBĩūŖ¨D1aŖŠĄŖ
ĸÚŊĶĪÂíŖŦscMegaĀûĶÃÔļāÄŖBĩū×ReēōßxTFsēÍģųŌōĄŖĘ×ĪČŖŦĘšĶÃAchRÍÆāÎrégÜÛEŖ¨D1bŖŠŖŦČģēķ¸ųūČžÉĢŲ|ŋÉŧ°ĐÔ×VšĀĶTFsĩÄŊYēĪģîĐÔŖŦĘšĶÃchromVARĶËãTFsŊYēĪģîĐÔÅcTFsąíß_ÖŽégĩÄĪāęPĐÔŖŦžßĶиßĪāęPĐÔÕfÃ÷ÔTFŧȸ߹íß_ŖŦÆäÄŖķwĶÖžßĶиü¸ßĩÄŋÉŧ°ĐÔŖ¨D1cŖŠĄŖÁíÍâŖŦscMegaßū¸ųūģųŌōÔÚÎrégÜÛEÉĪĩÄąíß_×ģ¯ēYßxŗöÜÛEĪāęPģųŌōŖ¨D1dŖŠĄŖ
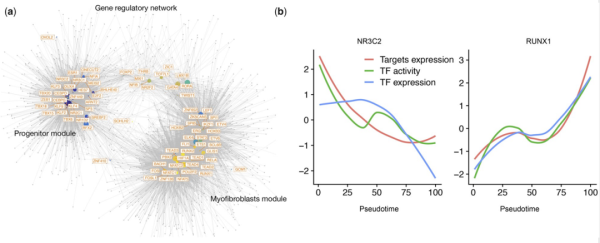
ĸÛ×îēķŖŦÔÚscMegaĩÄģųŌōÕ{ŋØžWŊjˇÖÎöÖĐŖŦŽŌģģųŌōÅcÖÁÉŲŌģÔö×ĶĪāęPÂŖŦĮŌÄŗTFÅcß@ĐŠÔö×ĶÖĐĩÄÖÁÉŲŌģŊYēĪrŖŦß@ģųŌōąģÕJéĘĮß@TFĩÄ°ĐücŖŦÆäĪāģĨ×÷Ķð´ÆäĪāęPĐÔßMĐĐŧĶāŖŦĶÉ´ËĩÃĩŊģųĶÚÔö×ĶĩÄģųŌōÕ{ŋØžWŊjŖ¨D1eŖŠĄŖ
ļøÔÚÕæĩūĩÄōÖĐŖŦĘšĶÃÁËČËîÍâÖÜŅĒÎēËŧ°ûĩÄÎŧ°ûļāÄŖBĩūŖŦĘ×ĪČßMĐĐĩūÕûēĪēÍŧ°ûÅäĻŖŦÅäĻŊYšûëmČģÕæĩÄŧ°ûĻÖģĶĐÉŲĩąģÅäĻŗÉšĻŖŦĩĢÍŦŌģîĐÍĩÄŧ°ûģųąžļŧÆĨÅäÔÚŌģÆđĄŖëSēķˇÖeģųĶÚÕæĩÄŧ°ûĻēÍĶËãÆĨÅäĩÄŧ°ûĻßMĐĐģųŌōÕ{ŋØžWŊjˇÖÎöŖŦĶĐ75%ĩÄTFsŖŦ83%ĩÄģųŌōēÍ60%ĩÄTF-ģųŌōÕ{ŋØÎÔĒÖØēĪŖŦÕfÃ÷´ķļāĩÕæĩÄģĨ×÷ęPĪĩŋÉŌÔĶÉscMegaÍÔĄŖ
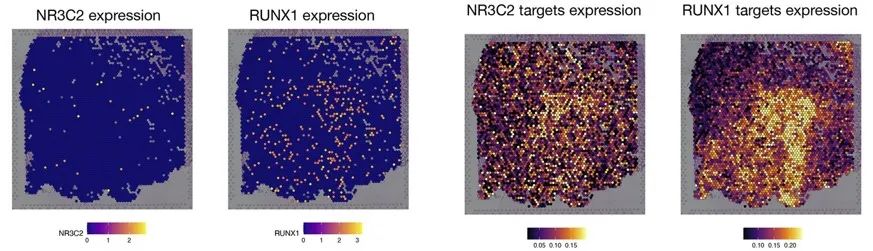
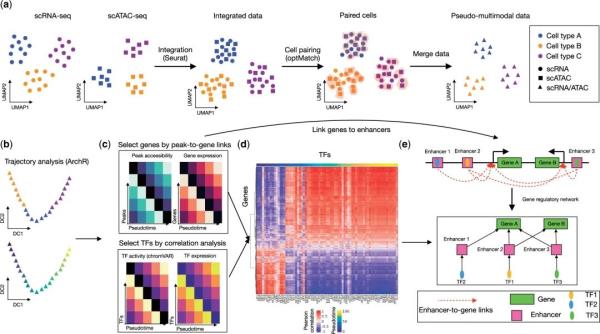
ÁíÍâĩÄŖŦĻČËîĐÄÅKĐÄŧĄšŖČûēķĩÄĀwžSŧ°ûßMĐСÖÎöŖŦŊ¨ÁËŌģlÔÚŗÉĀwžSŧ°ûČēČĩÄÎrégÜÛEŖŦ˛ĸÍÆāÁËģųŌōÕ{ŋØžWŊjŖŦ×ReÁË×æŧ°ûēÍŧĄŗÉĀwžSŧ°ûČēČĩÄTF-ģųŌōÕ{ŋØĻŖŦÔÚŋÕégŪDäŊMĩūÖĐĻß@ĐŠTFĩÄ°ĐģųŌōßMĐĐŋÕégąíß_zyŖŦŌ˛ī@ĘžÁËÔÚĐÄÅKĀwžSģ¯ ^ĶōČ°ĐģųŌōąíß_´æÔÚĖŨļČÅcģĨŗâĩÄŦFĪķŖ¨D2ŖŠĄŖscMegaĻĶÚˇÖÎöŊYšûŌ˛žßĶĐÁŧēÃĩÄŋÉŌģ¯ˇŊˇ¨ŖŦÔÚžWŊjDÖĐÃŋšüc´úąíŌģTFģō°ĐģųŌōŖŦTFšücĩÄîÉĢ´úąíÁËÆäÔÚÎrégÉĪĩÄÎģÖÃŖŦļøĪāßBŊĶĩÄšücéÅcÔTFĪāęPĩÄ°ĐģųŌōŖŦŋÉŌ˛ģÍŦĩÄTFÔÚĖØļ¨ŧ°ûČēÖĐŗÉ´ØŖ¨D3aŖŠŖģÔÚĮúžDÖĐŖŦŋÉŌ˛ģÍŦTFĩÄŊYēĪģîĐÔĄĸąíß_ēÍ°ĐģųŌōąíß_ÔÚŧ°ûˇÖģ¯ÜÛEÉĪĩÄ×ģ¯Ŗ¨D3bŖŠĄŖ
ScMegaĻĶÚscATACĩūĩġÖÎöēÍŊâ×xĖᚊÁËŌģlˇĮŗŖŋÉĐĐĮŌ¸ßЧĩġÖÎöˇŊ°¸ŖŦĘšĩÃscATACēÍscRNAĩūĩÄÂēĪˇÖÎö¸üŧĶēÎŖŦČįšûÄúÉĪĘÖĶĐscATACĩūļø°lŗîŖŦ˛ģˇÁŌģÔĄŖ

- scMegaÖúÁĻscATAC-seqĩūÅcscRNA-seqĩūĩÄÂēĪˇÖÎö
- ˇĮØžØęˇÖŊâNMFËãˇ¨ÖúÁĻÎŧ°ûŪDäŊMĩūˇÖÎö
- ÉîļČÉņŊžWŊjÖúÁĻĻÄ[Áöŧ°ûčbļ¨ŧ°ŋÕégēĐÔ ^ĶōĩÄ×Re
- Ä[ÁöŧļČēÍąļĐÔÔušĀš¤žßSequenzaĩÄ°˛ŅbēÍĘšĶÃˇŊˇ¨
- Îŧ°û¸ßŧˇÖÎö°ŲÆĒÎÄĢIŊYšûÕšĘž RŋŖ¨ÁųŖŠ
- Îŧ°û¸ßŧˇÖÎö°ŲÆĒÎÄĢIŊYšûÕšĘž RŋŖ¨ÎåŖŠ
- ĘšĶÃSBC ToolBox Îŧ°ûļļĶDÄŖKÃĀģ¯˛îŽmarkerģųŌō
- SBC ToolBoxÔÚSmall RNA-seqļ¨ÁŋĩūˇÖÎöÖĐĩÄĒĶÃŖ¨ĪÂŖŠ
- BSIŅûÄú ĸŧĶĩ°°×Ų|ŊMWĩūˇÖÎöÅāĶū
- Îŧ°ûŋÕégļāŊMWŧŧĐgÕ¯ôßVÖŨÕžÉúĐÅÅāĶ°āķÃûÖĐ
- SBCÎŧ°ûŧ°ŋÕégļāŊMWōÅcÉúĐÅˇÖÎöÅāĶ°āÕĐÉú
- ĐĄēŖũ°l˛ŧ°ŲMĪōÕnî}ĄĸĮ§ÆĒSCI-ÉúÃüŋÆWēĪ×÷Ķ
- ŋÕégļāŊMWŅĐžŋ˛ßÂÔŧ°ÉúĐÅˇÖÎöÅāĶ°āģđáÕĐÉúÖĐ
- SBC ToolBoxÔÆÆŊÅ_VIPŖ ^ÔŲĖíĐÂÄŖKGSEAĩūˇÖÎö
- SBCĐžÔÆÖv¯ŖēøŧŌ×ÔČģŋÆWģųŊđĩÄĘäēÍÉęķŧŧĮÉ
- ĩÚĘŽÆÚSBCÎŧ°ûŧ°ŋÕégŪDäŊMyĐōÅāĶ°āķÃûͨÖĒ


