
qPCR體系優化和常見問題解析大全
前言
聚合酶鏈式反應(PCR)是用于擴增特定DNA片段的分子生物學實驗技術。實時熒光定量PCR(以下簡稱qPCR)作為第二代PCR技術,自1996年推出以來,已經廣泛應用于基因表達分析、病原微生物檢測、動植物育種等許多研究領域,為了獲得最理想的檢測結果,qPCR從樣本采集、核酸提取、cDNA合成到上機檢測的流程有許多可以優化的參數。
qPCR實驗的工作流程首先需要確定研究的目的,根據實驗設計規劃好實驗分組、重復次數等細節。接下來分為樣本準備和引物探針驗證兩個重要的步驟。樣本準備主要是核酸提取逆轉錄等步驟,引物探針需要去測試特異性和效率。接下來需要使用qPCR儀來對樣品中的目的核酸進行擴增qPCR結束后根據實驗目的對目的核酸進行相對或者絕對定量。接下來講的qPCR體系優化會圍繞著這個流程展開。
1.樣本的采集與處理
首先,提前做好功課,了解樣本的不同分型,或者了解詳細的細胞分群。如果條件允許盡可能覆蓋所有的組織類型或者細胞類型。其次,盡可能增加樣本數量,也就是生物學重復,從而更客觀地反映生物變異程度。另外,qPCR實驗也需要有技術重復來降低誤差。
采樣是需要嚴格規劃的過程,比如材料的時效性、珍貴程度等,都要納入考量范圍。樣品要盡量新鮮,取樣盡可能快速。戴手套操作,防止污染。如果不馬上提取核酸,需要-80°C保存,并盡快處理。
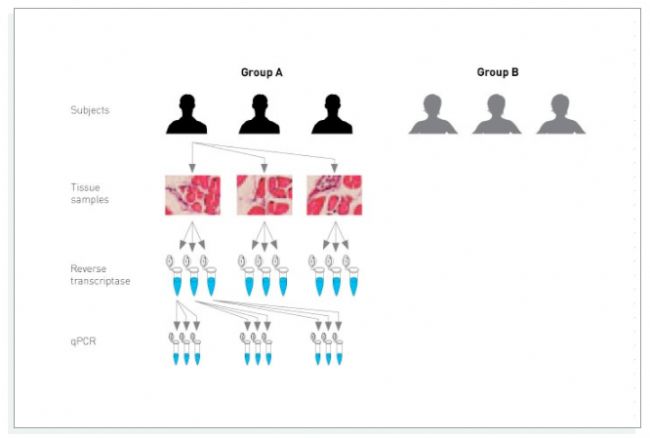
2.核酸的提取和檢測
模板的質量直接影響到檢測性能。核酸提取需要有效地將RNA或DNA從其他混合物中分離。RNA樣本中的污染物——基因組DNA、DNA結合蛋白、酚類化合物或在提取RNA過程中引入的外源雜質(如手套中的粉末)——都已被證明會抑制下游實驗,如逆轉錄和PCR擴增。核酸提取需要使用無菌無酶的試劑耗材,避免RNase或DNase污染,并對內源RNAse或DNAse進行有效抑制;多糖多酚樣品要考慮多糖多酚雜質的有效去除。低溫保存防止RNA或DNA降解。
降解或不純的RNA會限制逆轉錄反應的效率,降低產量。部分降解的RNA可能不能給出準確的基因表達結果。對于基因的定量,必須使用高質量的RNA,這意味著需要非常仔細地檢查RNA的濃度和質量。可采用高分辨率瓊脂糖凝膠檢測核酸質量和分光光度法(A260/A280=1.8和A260/A230=2.0)檢測核酸純度和濃度。
3.cDNA合成
RNA 質量對 cDNA 合成結果會產生重要影響。并且RNA 很脆弱,容易降解。為了保證 RNA 的完整性,我們需要非常注意,比如在冰上操作,用 RNase-free 的槍頭和離心管,減少操作時間等。在反應體系中加入 RNase 抑制劑也能有效防止 RNA 降解。
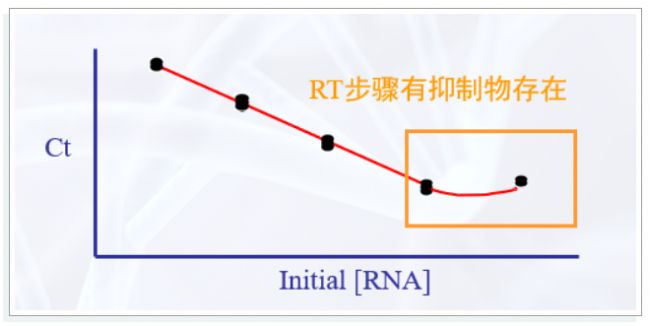
如何評價樣品中的雜質對逆轉錄的影響呢?可以梯度稀釋后繪制標準曲線,如果低濃度的樣品點數值偏大比較明顯,基本可以判定雜質影響顯著。
不同廠家的反轉錄試劑會有差異,對RNA中的雜質耐受程度也不同。逆轉錄酶在整個反轉錄體系中具有關鍵性影響。除了活性以外,逆轉錄酶的熱穩定性同樣很重要,在較高溫度下進行逆轉錄,能夠減少 RNA 的二級結構,增加逆轉錄的效率。
除了掌握 RNA 的完整性之外,反轉錄之前還需要對 RNA 濃度進行測定。一般反轉錄試劑盒會對上樣量有要求,建議 total RNA 上樣量小于 5 μg。超過這個范圍,會使反轉錄產物產生偏好性 (表達豐度高的基因優先被反轉錄) 而造成定量結果不準確。
逆轉錄出來的cDNA可以直接放在4°C保存,若長期不用,可分裝,然后-20°C保存。
4.qPCR方法的建立
① 定量方法
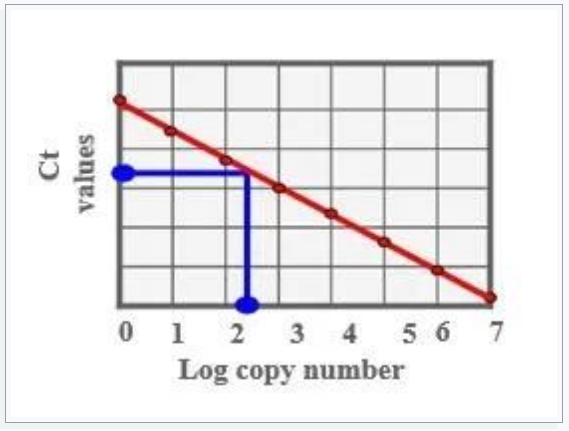
絕對定量:檢測起始模板數的精確拷貝數,需要標準品構建標準曲線。
標準品可以是純化的基因組DNA、質粒DNA或者體外轉錄RNA(cDNA),其作用是生成標準曲線,建立Ct值與濃度之間的線性關系。
標準品與待測樣品的PCR效率一致,且接近100%,與樣品的性質盡可能接近,與樣品相同的擴增條件(PCR體系、耗材、同一次擴增),大于或等于5個梯度稀釋的標準品。
相對定量:在一個樣本中,目的基因相對于內參基因的量的變化。
內參基因選擇建議篩選不少于三個內參基因來歸一化RT-qPCR數據。目的是消除外部樣品偏差,例如總RNA含量,RNA穩定性,酶效率或樣品裝載量的變化。
對候選的內參基因進行qPCR 實驗,得出Ct平均值以及 Ct值的標準偏差,選擇SD最小的基因作為實驗內參。可通過geNorm 、 BestKeeper 、 NormFinder、RefGenes 等工具來評估您的內參基因。
② 熒光標記方法
染料法:利用能與DNA雙鏈結合的染料來實現,如SYBR Green I。該染料在游離狀態下呈現微弱的熒光,一旦與雙鏈DNA的雙螺旋小溝結合,其綠色熒光增強約1000倍。因此其總的熒光強度與雙鏈DNA含量成正比,利用這一關系可以反映生成的PCR產物的量。
TaqMan熒光探針:是一種寡核苷酸探針,熒光基團連接在探針的5'末端,而淬滅劑則在3'末端。PCR擴增時在加入一對引物的同時加入一個特異性的熒光探針,探針完整時,報告基團發射的熒光信號被淬滅基團吸收; PCR擴增時, Tag酶的5'-3'外切酶活性將探針酶切降解,使報告熒光基團和淬滅熒光基團分離,從而熒光監測系統可接收到熒光信號,每擴增一條DNA鏈,就有一個熒光分子形成,實現了熒光信號的累積與PCR產物形成完全同步。常用的熒光基團是FAM,TET,VIC,HEX。
引物探針設計可以參考Gene π網站:https://www.gene-pi.com/item/primers-and-probes-2/
③ 引物擴增效率驗證
標準曲線是評估PCR擴增效率最可靠和穩定的一種方法,該方法涉及到制作一系列的樣品來控制目標模板的相對數量。最常用的是10倍梯度稀釋樣品,采用標準qPCR程序進行擴增獲得Cq值,最后根據各樣品濃度及相應的Cq值繪制標準曲線,得到線性方程Cq= -klgX0+b,擴增效率E=10(-1/k)-1。利用qPCR進行定量分析時,要求擴增效率范圍在90%-110%(3.6>k>3.1)。
④ 反應體系優化
▶ 根據儀器類型,選擇合適的耗材和qPCR試劑。
▶ 每對引物先進行預實驗,確定特異性以及最適濃度。
▶ 配置不同的PCR反應體系,選擇每個組分合適的濃度。
▶ 設置溫度梯度測試引物最合適的退火溫度。
▶ 實驗設置NTC、NRT、 NEG和POS等對照組,來監控實驗體系或污染。
實時熒光定量PCR常見問題分析
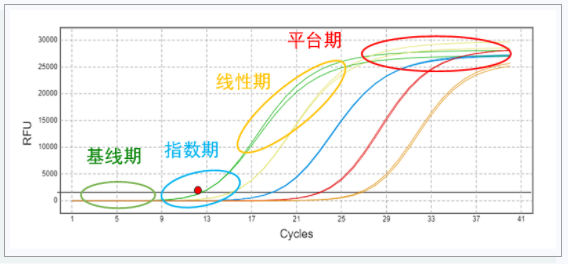
1.可疑的擴增曲線
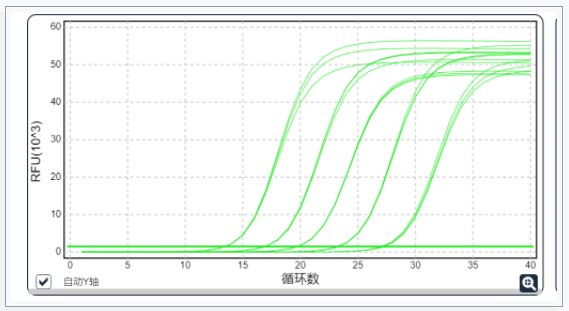
真正的擴增曲線,有特征的形狀:首先背景信號,然后是三個增長階段(指數增長期、線性增長期和平臺期)。
如果不是同時具有特征性的三個增長階段,沒有典型的指數增長期,那就不存在擴增。
平臺期很低也是常見的異常擴增曲線。可能是模板的濃度太低。通常如果模板的起始濃度太低, 反應體系中會形成大量的引物二聚體。大量引物二聚體的形成使得引物很快消耗完,從而造成擴增曲線的平臺期很低。這種情況可通過調整引物和模板的比例。
2.異常的熒光信號
NTC出現熒光信號---引物二聚體形成或氣溶膠污染,查看熔解曲線是否為單一峰。
3.擴增效率過高或過低
過低的擴增效率(<90%)可能存在的原因:
▶ 移液器校準不良或移液技術差。
▶ 不正確的稀釋導致標準曲線出現錯誤。
▶ 引物設計不好或擴增子具有二級結構。
▶ 標準曲線動態范圍太小。
▶ Taq酶無活性或活性降低。
▶ 樣品抑制。
過高的擴增效率(> 110%)可能存在的原因:
▶ 移液器校準不良或移液技術差。
▶ 不正確的稀釋導致標準曲線出現錯誤。
▶ 引物二聚體或非特異性擴增。
▶ 標準曲線動態范圍太小。
▶ 基因組DNA污染。
4.重復性差
為精確定量,對每個樣品都要做重復實驗,復孔之間的Ct值不應超過0.5,標準偏差不大于0.2,這樣,實驗結果就有很好的精確度。
造成重復性差的原因:
▶ 加樣誤差(操作或者加樣器導致)。
▶ 沒有將試劑和樣品充分混勻。
▶ 低拷貝的目的片段→泊松分布。
▶ 基線閾值設定不合理。
Cielo™實時熒光定量PCR系統
Harness of the power of qPCR

☑ 數據可靠性:連續1000次實驗后,結果高度一致。
☑ 應用靈活性:提供多種qPCR應用分析。
☑ 流程智能化:中英文用戶界面,觸控操作,可多機聯用。
☑ 在線便捷性:主機可獨立運行qPCR程序,數據可USB、Wi-Fi等網絡傳輸。



